![]() October 2024
October 2024
|
Hypertrophic cardiomyopathy (HCM) affects the heart muscle and can present at any age. HCM is usually diagnosed by echocardiogram. Symptoms range from mild shortness of breath on exertion to sudden cardiac death, often in young athletes. Early identification of HCM provides the best opportunity to implement clinical and lifestyle management strategies, potentially reducing mortality. HCM should be considered in cases of sudden death in young people. Up to 60% of cases will have an identifiable genetic cause. HCM is most often inherited in an autosomal dominant pattern. Genetic testing is recommended for:
All first-degree relatives of patients with a clinical diagnosis of HCM should have baseline clinical screening with echocardiography and a resting ECG, regardless of age or genetic test results. Ongoing clinical screening should be individualized. Treatment options depend on risk stratification and symptoms. Non-pharmacological management includes patient education (e.g. avoidance of vasodilators and Valsalva manoeuvre, importance of hydration). Other treatments may include management of heart failure and atrial fibrillation, implantable cardioverter defibrillators, surgery, medications (e.g. b-blockers, calcium channel blockers) that decrease afterload. Current evidence suggests the potential benefit of exercising and a more permissive approach to exercise for all stable HCM patients. Those who wish to engage in vigorous exercise such as competitive sports should be referred to a HCM expert. |
Download the PDF here. For a tool to be used at the point of care click here. For a more concise version see the GECKO on the run. For comprehensive guidelines on diagnosis, management and treatment see the 2024 Canadian Cardiovascular Society publication.
Jump ahead to a section:
What is hypertrophic cardiomyopathy?
What causes hypertrophic cardiomyopathy?
How is hypertrophic cardiomyopathy diagnosed?
Genetics and hypertrophic cardiomyopathy
Who should be offered genetic testing?
What are the benefits of genetic testing?
What do the genetic test results mean?
HCM resources for patients and clinicians
What is hypertrophic cardiomyopathy?
Hypertrophic cardiomyopathy (HCM) is a condition in which the myocardium is thickened, and the myocytes are fibrotic and disorganized. HCM is a major cause of morbidity and mortality, including exertional symptoms, heart failure, atrial fibrillation (AF), stroke, and ventricular arrhythmias, which can sometimes result in sudden cardiac arrest or death.1 Hypertrophy can be septal, apical, or concentric. Asymmetrical ventricular hypertrophy is the most common. Obstructive HCM occurs when left ventricular outflow is compromised. Symptoms can include dyspnea, chest pain, palpitations, syncope and, in some cases, sudden cardiac death even in the absence of marked hypertrophy.
HCM is common. The prevalence is estimated to be about 1 in 500, but possibly as common as 1 in 200 when accounting for asymptomatic carriers of pathogenic gene variant(s).2,3
The onset and presentation of HCM is highly variable. This can also be described as variable expressivity, where the presentation of the condition varies from person to person, even within the same family or between those with the same underlying genetic cause. HCM can present from infancy to late adulthood. HCM may be isolated or related to a broad spectrum of causes such as inborn errors of metabolism, various genetic syndromes, and neuromuscular disorders.2 Individuals presenting before the age of one year have the most diverse etiologies and poorest outcomes.4
HCM may account for 13-50% of all sudden cardiac deaths [SCD]2, although risk of SCD is about 0.5-1% in the general HCM population.5 Among patients with HCM, younger patients are at higher risk for SCD than older patients.6
What causes hypertrophic cardiomyopathy?2,5,6
The etiology of HCM is variable but most causes are genetic and follow an autosomal dominant pattern of inheritance with variable expressivity. 35-60% of HCM cases will have an identifiable genetic cause.2,6 The detection rate of genetic testing is higher in those with a family history of the same condition. In some families there can be multiple genetic variations contributing to HCM.
About 5 to 10% of cases of HCM are acquired, due to environmental factors like hypertension, valve disease, aortic stenosis, or athlete’s heart. Less than 1% HCM cases occur as part of a genetic syndrome where extracardiac features would be observed, such as Noonan syndrome or Fabry disease. The remaining cases have an unknown etiology.
How is hypertrophic cardiomyopathy diagnosed?
Many patients with HCM are asymptomatic. The clinical evaluation for HCM can be prompted by a positive family history of HCM, the presence of symptoms such as a cardiac event, the detection of a heart murmur during a physical exam, abnormal findings on an echocardiogram conducted for other reasons, or irregularities on a 12-lead ECG (e.g. repolarization abnormalities, arrhythmias). Individuals with HCM typically exhibit a systolic murmur, a pronounced apical point of maximal impulse, an abnormal carotid pulse, and the presence of a fourth heart sound.6
Syncope with exercise is a warning symptom of HCM and other potentially heritable heart problems in young athletes and should be thoroughly investigated.1
Echocardiography is the main imaging evaluation for HCM.1,2,6 Cardiac MRI, also called cardiovascular magnetic resonance (CMR), allows for detailed analysis of the morphological abnormalities present in HCM. CMR should be considered in all patients with suspected HCM and is complementary to echocardiography.1 CMR may also aid risk stratification.7 Diagnostic criteria are outlined below where A and B are fulfilled.
Genetics and hypertrophic cardiomyopathy
The most common genes associated with HCM are: MYBPC3, MYH7, TNNI3, TNNT2, ACTC1, MYL2, MYL3, TPM1. Over 1,500 genetic variants have been identified. The majority of HCM-related genes are inherited in an autosomal dominant manner and are responsible for sarcomere or sarcomere-related proteins.
Typically, an individual with HCM will have one pathogenic (P) or likely pathogenic (LP) variant in one HCM gene, however, some individuals will have P/LP variants in more than one HCM-related genes where each variant contributes modestly to HCM risk (polygenic).1
Less than 1% of individuals who have HCM will have extracardiac features that are part of a genetic syndrome.2 Some examples are:
- Noonan syndrome/RASMARK pathway (genes: PTPN11, RAF1, SOS1, others), autosomal dominant inheritance
- Fabry disease (gene: GLA), X-linked inheritance
- Inborn errors of metabolism, autosomal recessive inheritance
Genetic testing is performed with a panel where multiple genes, sometimes 12 or more, are analyzed at the same time. HCM panels may not include genes associated with syndromic presentation. HCM panels can also differ between laboratories. Consult your local genetics specialist to discuss if the available testing is appropriate for your patient (e.g. testing for the familial gene variant is possible.) Results can be complicated and require specialized interpretation by genetics experts.
Who should be offered genetic testing?
Genetic testing must start with an affected individual. The most informative person to begin genetic testing is the youngest, affected person. This is the person most likely to carry a pathogenic (P) or likely pathogenic (LP) gene variant. Testing is not recommended in unaffected persons, unless there is a known P/LP gene variant in the family.
Genetic testing is recommended for:1
- All patients with a confirmed or suspected clinical diagnosis of HCM.
- Patients with a known pathogenic/likely pathogenic HCM-genetic variant in their family.2
- Deceased individuals with a suspected inherited cardiac condition as part of an autopsy.2
What are the benefits of genetic testing?
|
Genetic testing for HCM can help with:
|
Genetic testing:
|

What do the genetic test results mean?
The detection rate of genetic testing for HCM is approximately 60%, which means that 40% of individuals with HCM will not receive an informative (positive) result. Apart from those with true negative results, all first-degree relatives should have at least baseline echocardiographic and ECG screening regardless of age. Individuals who opt not to have testing for a known familial gene variant should have periodic cardiology assessment.
Test results fall into three categories:
Positive: causative pathogenic (P) or likely pathogenic (LP) variant detected in an HCM-related gene
- This confirms a genetic etiology and a diagnosis of HCM in cases of suspected HCM.
- These results can be used to initiate cascade testing of relatives. First degree relatives (children, parents, siblings) have a 50% chance to also carry the familial P/LP gene variant and to be at risk for HCM.
- All first-degree relatives should be offered
- Asymptomatic individuals who are positive for a familial gene variant would require periodic cardiology assessment.
- While results are not likely to predict long term outcomes, positive results can indicate earlier onset of disease and worse outcomes.1
- Limited data are available to determine the ideal frequency for clinical screening and the appropriate age at which screening should be discontinued. A frequency of every 1-2 years until the age of 20 and every 3-5 afterwards for echocardiographic screening is reasonable.1
Negative
Uninformative: no causative pathogenic variant detected in any HCM-related genes tested
- Heritable HCM is neither confirmed nor ruled out.
- Genetic testing cannot be offered to unaffected relatives.
- For individuals with HCM in whom no genetic cause is identified, updates to genetic panels or technology should be reviewed every 3-5 years, especially for families with multiple affected individuals1
True negative: the familial pathogenic/ likely pathogenic variant is not detected
- This is where genetic testing is offered to an unaffected relative after the causative pathogenic gene variant has been identified.
- The individual does not have the familial condition and is not at increased risk to develop HCM.
- This individual’s offspring would not have increased risk for HCM.
Variant of uncertain significance (VUS): a variant in an HCM-related gene is detected, but there is insufficient evidence to determine if it is truly associated with disease. (It is possible for more than one VUS to be detected.)
- The genetics team may offer testing to other relatives affected with HCM to see if the variant is present in those with disease and absent in unaffected relatives (segregation study).
- This result will not affect diagnosis or management.
- Over time, additional information about the VUS may become available, 10%-15% of variants are reclassified (as benign or pathogenic) on follow-up. Re-referring to genetics can be considered in approximately 5 years or as recommended by Genetics.1
Visit the GECKO site to find more resources on genetic test results including a point of care tool.
Surveillance and management
Early recognition of HCM can enable effective treatment of symptoms in most patients.6 Risk stratification and treatment options should be assessed by an expert in HCM.
Screening
Figure 1 depicts a general approach to clinical and genetic screening of patients and relatives.
Clinical screening should be individualized. Limited data are available to determine the ideal frequency for clinical screening and the appropriate age at which screening should be discontinued. A frequency of every 1-2 years until the age of 20 and every 3-5 afterwards for echocardiographic screening is reasonable.1 The yield of clinical screening in families with uninformative genetic test results, especially if HCM is mild and diagnosed at an old age in a single relative, is likely to be relatively low.
|
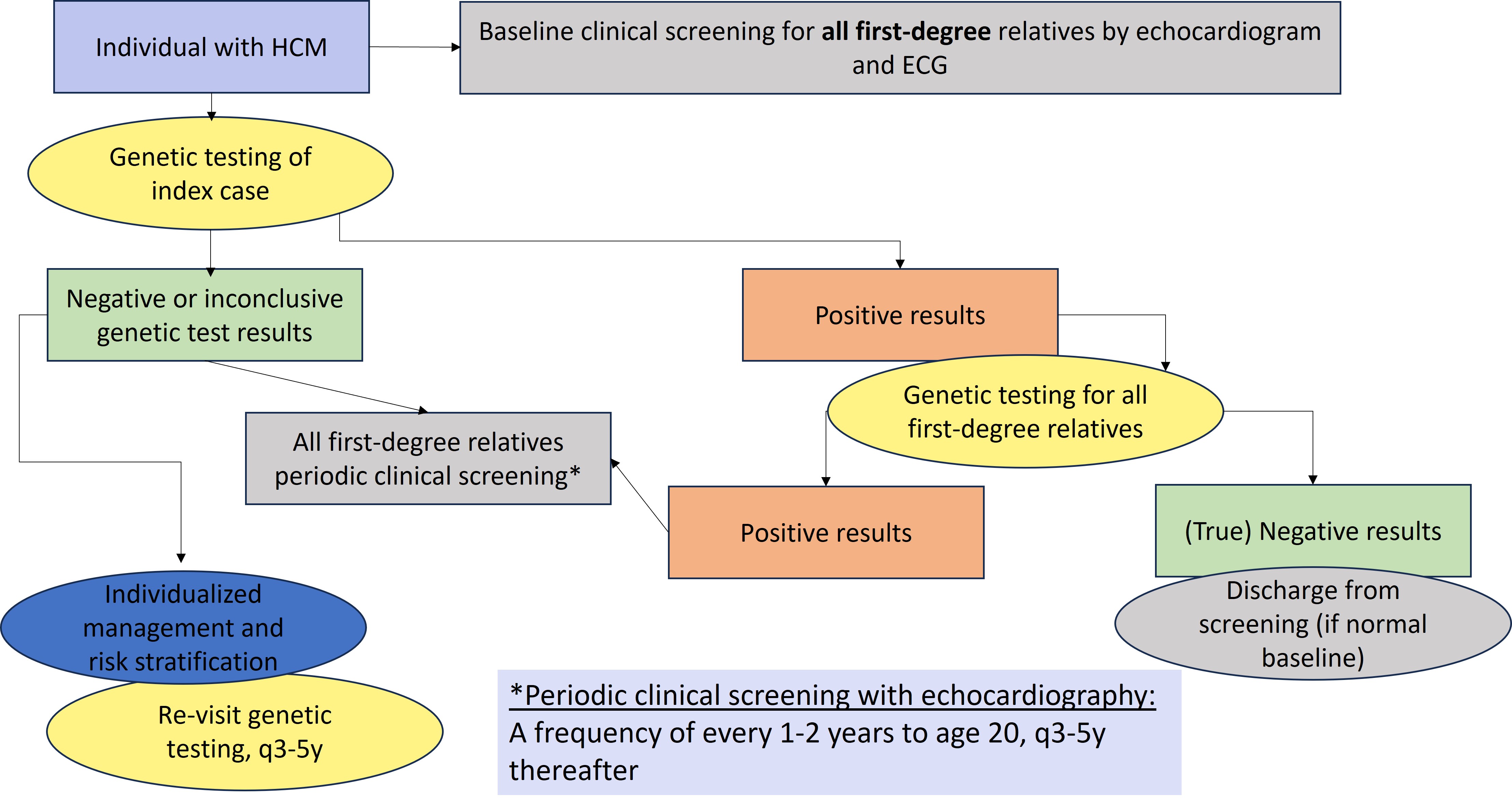
Figure 1. General approach to genetic testing and family screening. ECG, electrocardiogram; HCM, hypertrophic cardiomyopathy; P/LP, pathogenic or likely pathogenic.1
Tips for clinical screening from the Canadian Cardiovascular Society (2024)1:

How is HCM treated?
Treatment options depend on risk stratification and symptoms. Non-pharmacological management includes patient education (e.g. avoidance of vasodilators, Valsalva manoeuvre, importance of hydration). Other treatments may include management of heart failure and atrial fibrillation, implantable cardioverter defibrillators, surgery, medications (e.g. b-blockers, calcium channel blockers). See the recent Canadian Cardiovascular Society (2024) publication1.
Risk stratification
Risk stratification is a major treatment initiative in lowering mortality. This is used to identify patients with HCM who are at the greatest risk of sudden cardiac death (SCD).6 It allows for more accurate identification of patients suitable for implantable cardioverter defibrillators (ICDs) as primary prevention for SCD.5 The decision for ICD should be part of shared decision-making between a patient and their cardiologist. Risk stratification relies on standalone high-risk clinical features and estimated high-risk based on validated calculators (Table 1).1 (CCS) The HCM risk-SCD risk model is the most accurate calculator to date with some validation studies, although not valid in all populations.5
Table 1. Clinical and estimated risk factors for SCD in those with HCM.
The risk of SCD is highest in middle-aged patients and young adults versus those of >60 years6. Each of the risk factors listed below are major risk factors for SCD. Consideration of primary prevention with ICD may be reasonable in those with >1 risk factor(s). More details can be found in Table 8 of the recent Canadian Cardiovascular Society publication.
|
Massive left ventricle hypertrophy |
Maximal wall thickness >30 mm |
|
Unexplained syncope |
Recent history (events more than 5 years ago appear to not be relevant); Assumed not to be of vasovagal etiology (neurocardiogenic) or due to left ventricle obstruction |
|
Left ventricle ejection fraction < 50% |
As measured by echocardiogram or cardiac MRI |
|
Apical aneurysm |
A discrete thin-walled dyskinetic or akinetic segment of the most distal part of the left ventricle |
|
Extensive fibrosis defined as late gadolinium enhancement (LGE) involving >15% of the left ventricle |
Diffuse and extensive LGE, representing fibrosis |
|
Strong family history for SCD |
One or more first- or second-degree relative(s), multiple tertiary relatives |
|
Non-sustained ventricular tachycardia (NSVT) |
In adults who also have high-risk features (e.g. fast, long duration) |
|
Positive genetic test |
Carrier of one of more likely pathogenic or pathogenic gene variants in HCM-related gene(s) associated with SCD |
|
HCM Risk-SCD risk model score of >4% within 5 years |
HCM Risk-SCD calculator: https://qxmd.com/calculate/ calculator_303/hcm-risk-scd |
|
Children |
|
|
Children with massive left ventricle hypertrophy, any NSVT or with high-risk of arrythmia as calculated by validated calculator |
· An absolute or z-score threshold for wall thickness has not been established; however, a maximal wall that corresponds to a z-score >20 (and >10 in conjunction with other risk factors) appears reasonable. (AHA) · Presence of any NSVT in children · Children with high risk of arrhythmic events as calculated by: o Precision Medicine for Cardiomyopathy (PRIMaCY): https://primacycalculator.com o HCM Risk-Kids:10 https://hcmriskkids.org |
Exercise and HCM
Traditionally those with HCM have exercise restrictions. There is more current evidence suggesting the potential benefit of exercising and a more permissive approach is recommended.1 For all stable HCM patients, moderate exercise is recommended. Those who wish to engage in vigorous exercise such as competitive sports should be referred to specialised HCM expert.1
References
[2] Care M, Chauhan V, Spears D. Genetic Testing in Inherited Heart Diseases: Practical Considerations for Clinicians. Curr Cardiol Rep. 2017 Aug 16;19(9):88.
[3] Semsarian C, Ingles J, Maron MS, Maron BJ. New perspectives on the prevalence of hypertrophic cardiomyopathy. J Am Coll Cardiol. 2015 Mar 31;65(12):1249-1254.
[4] Colan SD, Lipshultz SE, Lowe AM, et al. Epidemiology and case-specific outcomes in Hypertrophic Cardiomyopathy in children: Findings from the Pediatric Cardiomyopathy Registry. Circulation 2007; 115: 773–81.
[5] Cooper RM, Raphael CE, Liebregts M, et al. New Developments in Hypertrophic Cardiomyopathy. Can J Cardiol. 2017 Oct;33(10):1254-1265.
[6] Ommen SR, Mital S, Burke MA, et al. 2020 AHA/ACC Guideline for the Diagnosis and Treatment of Patients With Hypertrophic Cardiomyopathy: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. J Am Coll Cardiol. 2020 Dec 22;76(25):e159-e240.
HCM resources for patients and clinicians:
- The Canadian Sudden Arrhythmia Death Syndromes (SADS) Foundation website http://www.sads.ca/
- Hearts in Rhythm Organization (HiRO) https://hiro.heartsinrhythm.ca/
- Genome BC. Cascade screening for hypertrophic cardiomyopathy https://vimeo.com/826986524/7969433f18
Authors: S Morrison MS CGC, JE Allanson MD FRCPC, S Walji MD CCFP MPH, JC Carroll MD CCFP
Special thanks to A Rideout MS CCGC CGC

