![]() October 2024
October 2024
|
Hypertrophic cardiomyopathy (HCM) affects the heart muscle and can present at any age. HCM is usually diagnosed by echocardiogram. Symptoms range from mild shortness of breath on exertion to sudden cardiac death, often in young athletes. Early identification of HCM provides the best opportunity to implement clinical and lifestyle management strategies, potentially reducing mortality. HCM should be considered in cases of sudden death in young people. Up to 60% of cases will have an identifiable genetic cause. HCM is most often inherited in an autosomal dominant pattern. Genetic testing is recommended for:
All first-degree relatives of patients with a clinical diagnosis of HCM should have baseline clinical screening with echocardiography and a resting ECG, regardless of age or genetic test results. Ongoing clinical screening should be individualized. Treatment options depend on risk stratification and symptoms. Non-pharmacological management includes patient education (e.g. avoidance of vasodilators and Valsalva manoeuvre, importance of hydration). Other treatments may include management of heart failure and atrial fibrillation, implantable cardioverter defibrillators, surgery, medications (e.g. b-blockers, calcium channel blockers) that decrease afterload. Current evidence suggests the potential benefit of exercising and a more permissive approach to exercise for all stable HCM patients. Those who wish to engage in vigorous exercise such as competitive sports should be referred to a HCM expert |
Download the PDF here. Check out the more comprehensive GECKO Deep Dive and the point of care tool.
For comprehensive guidelines on diagnosis, management and treatment see the 2024 Canadian Cardiovascular Society publication.
What is hypertrophic cardiomyopathy?
Hypertrophic cardiomyopathy (HCM) is a common (1 in 500) autosomal dominant condition in which the myocardium is thickened, and the myocytes are fibrotic and disorganized. HCM is a major cause of morbidity and mortality, including exertional symptoms, heart failure, atrial fibrillation (AF), stroke, and ventricular arrhythmias, which can sometimes result in sudden cardiac arrest or death.1 The onset and presentation of HCM is highly variable even within families. HCM may account for 13-50% of all sudden cardiac deaths [SCD]2, although risk of SCD is about 0.5-1% in the general HCM population.5 Among patients with HCM, younger patients are at higher risk for SCD than older patients.6
What causes hypertrophic cardiomyopathy?2,5,6
The etiology of HCM is variable but most causes are genetic and follow an autosomal dominant pattern of inheritance with variable expressivity. 35-60% of HCM cases will have an identifiable genetic cause.2,6 The detection rate of genetic testing is higher in those with a family history of the same condition. In some families there can be multiple genetic variations contributing to HCM.
About 5 to 10% of cases of HCM are acquired, due to environmental factors like hypertension, valve disease, aortic stenosis, or athlete’s heart. Less than 1% HCM cases occur as part of a genetic syndrome where extracardiac features would be observed, such as Noonan syndrome or Fabry disease. The remaining cases have an unknown etiology.
How is hypertrophic cardiomyopathy diagnosed?
Syncope with exercise is a warning symptom of HCM and other potentially heritable heart problems in young athletes and should be thoroughly investigated.1
Echocardiography is the main imaging evaluation for HCM.1,2,6 Cardiac MRI, also called cardiovascular magnetic resonance (CMR), allows for detailed analysis of the morphological abnormalities present in HCM. CMR should be considered in all patients with suspected HCM and is complementary to echocardiography.1 CMR may also aid risk stratification.7 Diagnostic criteria are outlined below where A and B are fulfilled.
|
Who should be offered genetic testing?
Genetic testing must start with an affected individual. The most informative person to begin genetic testing is the youngest, affected person. This is the person most likely to carry a pathogenic (P) or likely pathogenic (LP) gene variant. Testing is not recommended in unaffected persons, unless there is a known P/LP gene variant in the family.
Genetic testing is recommended for:1
- All patients with a confirmed or suspected clinical diagnosis of HCM.
- Patients with a known pathogenic/likely pathogenic HCM-genetic variant in their family.2
- Deceased individuals with a suspected inherited cardiac condition as part of an autopsy.2
What are the benefits of genetic testing?
|
Genetic testing for HCM can help with:
|
Genetic testing:
|

Surveillance and management
Early recognition of HCM can enable effective treatment of symptoms in most patients.(AHA) Risk stratification and treatment options should be assessed by an expert in HCM.
Screening
Figure 1 depicts a general approach to clinical and genetic screening of patients and relatives.
Clinical screening should be individualized. Limited data are available to determine the ideal frequency for clinical screening and the appropriate age at which screening should be discontinued. A frequency of every 1-2 years until the age of 20 and every 3-5 afterwards for echocardiographic screening is reasonable.1 The yield of clinical screening in families with uninformative genetic test results, especially if HCM is mild and diagnosed at an old age in a single relative, is likely to be relatively low.
- First-degree relatives of patients with a clinical diagnosis of HCM should have baseline clinical screening with echocardiography and a resting ECG, regardless of age and genetic test result.
- In families where a causative genetic variant has been identified (positive results)
- relatives who do not carry the variant (true negative results) can be discharged from follow-up, if they have normal baseline clinical screening.
- Relatives who carry the familial causative gene variant (positive results) should have periodic clinical screening (as above)
- First-degree relatives of those with HCM but no causative genetic variant (uninformative results or variant(s) of uncertain significance) should have periodic clinical screening
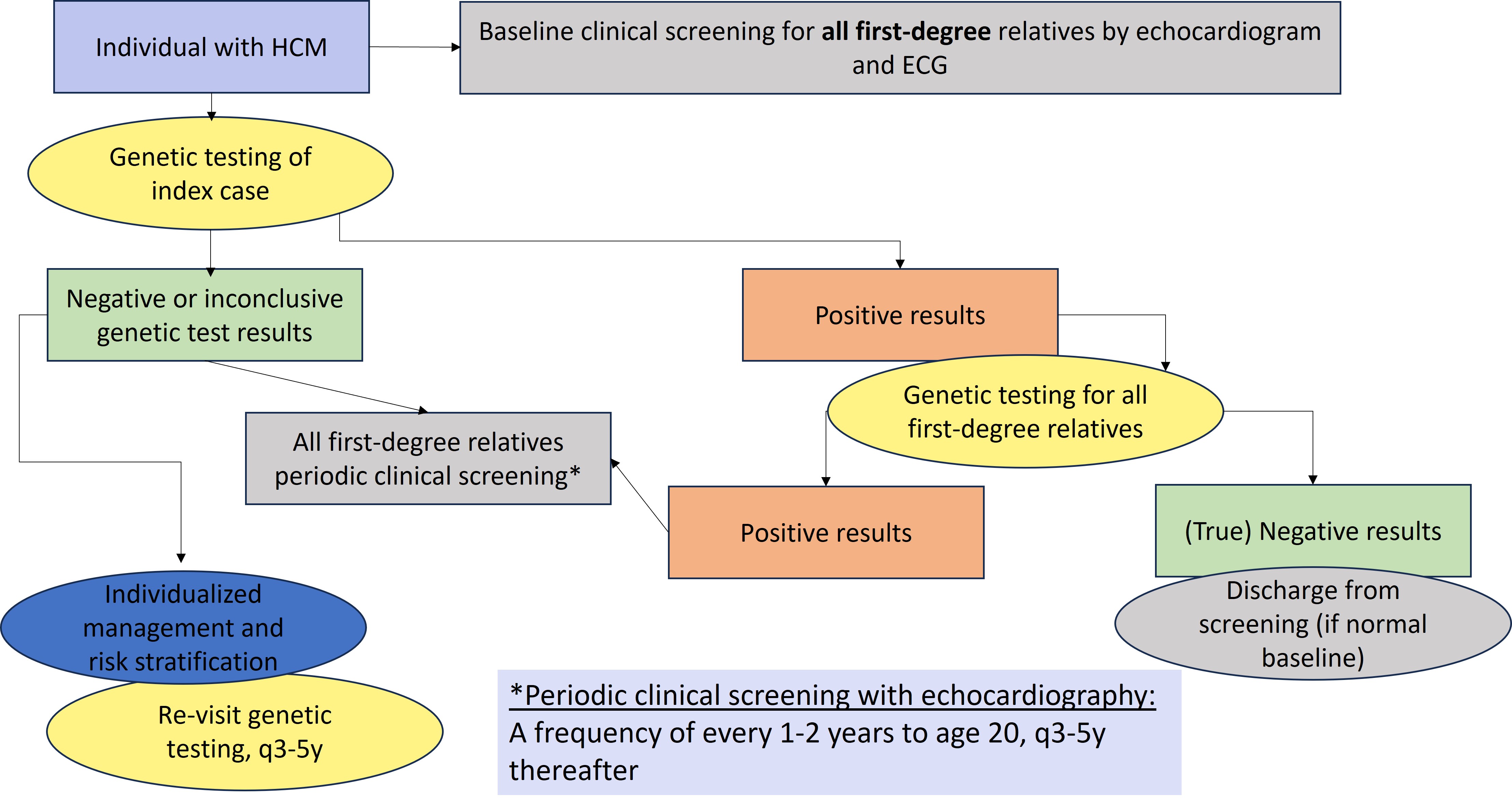
Figure 1. General approach to genetic testing and family screening. ECG, electrocardiogram; HCM, hypertrophic cardiomyopathy; P/LP, pathogenic or likely pathogenic.1
Key reference:
Crean AM, Adler A, Arbour L, et al. Canadian Cardiovascular Society Clinical Practice Update on Contemporary Management of the Patient With Hypertrophic Cardiomyopathy. Can J Cardiol. 2024 Sep;40(9):1503-1523. Full reference list can be found in the GECKO Deep Dive.
HCM resources for patients and clinicians:
- The Canadian Sudden Arrhythmia Death Syndromes (SADS) Foundation website http://www.sads.ca/
- Hearts in Rhythm Organization (HiRO) https://hiro.heartsinrhythm.ca/
- Genome BC. Cascade screening for hypertrophic cardiomyopathy https://vimeo.com/826986524/7969433f18
Authors: S Morrison MS CGC, JE Allanson MD FRCPC, S Walji MD CCFP MPH, JC Carroll MD CCFP
Special thanks to A Rideout MS CCGC CGC

