
Bottom line: Hereditary breast and ovarian cancer predisposition refers to an inherited increased lifetime risk to develop breast and/or ovarian cancer and accounts for about 5-15% of these cancers. The actual cancer risk depends upon the gene involved as well as personal and family history factors. Primary care practitioners (PCPs) have a role in identifying those who are at increased risk to carry a pathogenic/likely pathogenic (P/LP) variant in a gene associated with increased cancer risk, as well as facilitating familial testing when there is a known genetic predisposition to cancer. Genetic testing may be considered where there is a personal and/or family history of:
- A known P/LP variant in a gene associated with hereditary cancer.
- Multiple relatives (>3) with the same or related cancer diagnoses, on the same side of the family (e.g. breast and ovarian cancer).
- Multiple cancer diagnoses in one individual (e.g. bilateral breast cancer, breast and ovarian cancer)
- young age at diagnosis (<50y).
- High risk cancer diagnosis (e.g. triple negative breast cancer, high-grade metastatic castrate resistant prostate cancer)
- Rare/unusual cancer diagnosis (e.g. male breast cancer).
- At-risk ancestry (e.g. Ashkenazi Jewish) with a history of cancer.
PCPs also have a role in coordinating high-risk screening (e.g. MRI) and referring for specialist consultation for consideration of risk-reducing management and personalized screening recommendations.
Jump to a section or Ctrl + F to find what you are looking for
What is hereditary breast and ovarian cancer?
Who to consider referring for a genomic assessment for hereditary breast and ovarian cancer?
What do the genetic test results mean?
What are the benefits and considerations of genetic testing?
What are the cancer risks associated with an inherited predisposition?
Gene specific cancer surveillance and management recommendations
Reproductive considerations (family planning)
Related point of care tools and resources
What is hereditary breast and ovarian cancer?
All cancer is genetic, but not all cancer is inherited. This means that all cancer is the product of pathogenic changes (what used to be called mutations) in one or more genes that result in uncontrollable cell growth. However, most of these genetic changes are acquired and not inherited from a parent. In fact, only about 5-10% of breast cancer and about 15% of ovarian cancer is thought to be the result of an inherited pathogenic/likely pathogenic (P/LP) variant in a cancer predisposition gene.1 Often cancer predisposition genes have roles in regulating cell growth (tumor suppressor genes) or repairing damaged DNA. About 1 in 3 Canadians will develop cancer in their lifetime. All individuals have at least an average risk to develop cancer, but with additional risk factors (Figure 1), including an inherited risk, some Canadians have an increased lifetime risk of cancer.
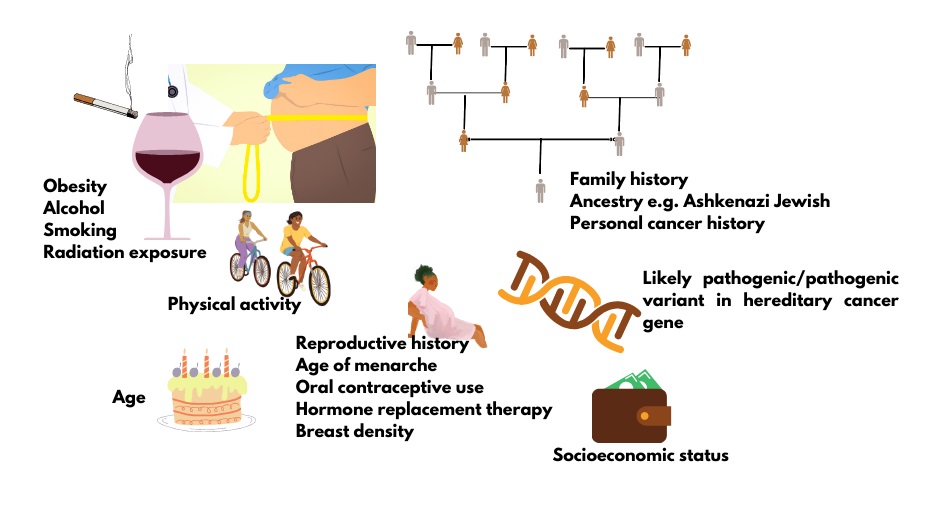
Figure 1. Factors that contribute to breast cancer risk.
The genes BRCA1 and BRCA2 account for about half of the hereditary breast and ovarian cancer families. Individuals with P/LP variants in these genes have an increased lifetime risk for not only breast and ovarian cancer, but also for contralateral breast, prostate, pancreatic cancers and more. Other high-risk genes (e.g. PALB2, PTEN) and moderate risk genes (e.g. CHEK2, ATM) account for most of the rest. Not every family will have an identifiable genetic etiology to explain their familial cancer. Current thought is these familial cancers may be the result of a combination of one or more moderate risk genetic variants and shared environmental factors.
Individuals with an inherited predisposition to cancer have a significantly increased risk for breast (2-6x) and/or ovarian (10-30x) cancer. The actual risk depends upon which gene has a P/LP variant as well as personal and family history factors. Not every individual with an inherited predisposition will develop cancer (reduced penetrance) and not every individual in a single family will develop the same cancer or have the same age of onset (variable expressivity). Because of the overlap in clinical presentation (genetic heterogeneity) of many hereditary cancer predisposition conditions and with advances in genomic sequencing, genetic testing by multi-gene panel is more efficient and cost-effective than testing one or two genes at a time. This resource will explore hereditary breast and ovarian cancer predisposition and the role of primary care practitioners.
Who to consider referring for a genomic assessment for hereditary breast and ovarian cancer?
Genomic centres across Canada have their own criteria for referral and genetic testing. Check out the GECKO site for how to contact your local genomics centre and for links to provincial breast cancer screening programs.
A first step to assessing those who would benefit from a genetic assessment is eliciting a family history. Validated tools exist2 to facilitate identifying those who are at an increased risk to have a hereditary predisposition to breast and ovarian cancer and there are general red flags to identify those at risk of any hereditary cancer predisposition (GECKO point of care tool: General hereditary cancer red flags). More specific red flags to interrogate when there is a suspicion for hereditary breast and ovarian cancer can be found in the GECKO point of care tool: hereditary breast and ovarian cancer red flags. Reach out to your local genetics provider by eConsult or phone if you have a question about referral, family history, screening or further assessment.
General personal/family history cancer predisposition considerations:
In general, these features raise the suspicion of a hereditary cancer predisposition:
- Multiple relatives (<3) with the same or related cancer diagnoses, on the same side of the family
- Cancers related to hereditary breast and ovarian cancer predisposition genes are:
- Breast
- Ovary
- Prostate
- Pancreas
- Young age at diagnosis (e.g. breast cancer <50y, pancreatic cancer <45y)
- Several affected generations
- Multiple cancer diagnoses in one individual (e.g. bilateral breast cancer, breast and ovarian cancer)
- High risk cancer diagnosis (e.g. triple negative breast cancer, high-grade metastatic castrate resistant prostate cancer)
- Rare/unusual cancer diagnosis (e.g. male breast cancer)
- Ancestry where there is a higher carrier frequency (e.g. The general population carrier frequency for pathogenic variants in BRCA1 and BRCA2 is 1 in 400 whereas in those of Ashkenazi Jewish ancestry the carrier frequency is 1 in 40)
- Cancers related to hereditary breast and ovarian cancer predisposition genes are:
Important pieces of information to clarify are:
- Cancer diagnosis
- Age at diagnosis
- Relation to patient
- Ancestry
In addition to clinical judgement and clinical criteria, genetics specialists may use risk models to estimate the probability an individual carries a pathogenic/likely pathogenic (P/LP) variant in a hereditary cancer gene as well as their lifetime cancer risks. These models consider personal health history factors such as age, height, weight, age at menarche, age at menopause, breast density, history of benign breast disease, as well as family history. The CanRisk Web Tool and the IBIS Breast Cancer Risk Evaluation Tool are validated models used by many genomic centres. While unaffected individuals are not typically offered genetic testing first in a family, if the probability is high enough (>5-10%) using an established risk model, testing may be considered when another more appropriate relative is not available.
With any suspected genetic condition, the best person in whom to begin genetic testing is always the person most likely to carry a P/LP variant in a candidate gene. With hereditary cancer predispositions, this person is typically the one diagnosed with cancer at the youngest age (e.g. breast cancer at age 40), the one diagnosed with multiple cancers (e.g. breast and ovarian cancer, bilateral breast cancer), or the one diagnosed with a rare cancer (e.g. male breast cancer).
If a P/LP variant in a hereditary cancer gene has already been identified in the family, all blood relatives are eligible for genetic testing for that variant. Unaffected individuals who are considering testing should be offered genetic counselling so that an informed decision is made about whether and when to pursue predictive testing. First degree relatives of carriers have a 50% chance to carry the same P/LP variant and to be at increased risk for the gene-associated cancers, and a 50% chance to not be a carrier. A negative result is reassuring but does not eliminate all cancer risk. Individuals who receive a negative result following familial variant testing are still advised to follow population-based cancer screening.
Treatment: Some individuals will be offered genetic testing not solely in the context of a suspicious hereditary cancer risk but for the purpose of treatment planning e.g. qualification for medication such as poly (adenosine diphosphate [ADP]-ribose) polymerase (PARP) inhibitors. While provincial testing criteria vary, in general, genetic testing is recommended for any ovarian, pancreatic and metastatic prostate cancer diagnosis, and breast cancer diagnosed at a young age (<50 years).3
Screening: Even if a patient does not qualify for genetic assessment or testing, they may still be eligible for increased cancer screening based on family history. The most informative person in a family to refer to genetics is always the person most likely to carry a P/LP variant in a hereditary cancer gene (e.g. cancer diagnosis at the youngest age (e.g. breast cancer <50 years), multiple cancer diagnoses (e.g. breast and ovarian cancer, bilateral breast cancer), or rare cancer diagnosis (e.g. male breast cancer)). Once a P/LP variant has been identified in a family, all other relatives can have testing for the specific familial variant.
Private pay genetic testing: Some individuals may wish to pay out of pocket for genetic testing, for example if they do not qualify by provincial criteria but still would like genetic testing. Generally, this testing will not be arranged by genetic centres but maybe requested of primary care practitioners, be direct-to-consumer, or through private health clinics. Several companies offer genetic testing for hereditary cancer predispositions. When choosing a genetic testing company consider whether:
- Pre- and post-test genetic counselling is offered and delivered by a qualified and certified health professional (e.g. Canadian Board of Genetic Counselling, American Board of Genetic Counseling).
- The genetic testing laboratory and personnel are accredited/certified by appropriate governing bodies (e.g. Accreditation Canada Diagnostics, Clinical Laboratory Improvement Amendments (CLIA) certification or appropriate provincial licensure bodies for laboratories, board-certification of genetic counsellors and geneticists).
- Testing analyses the whole gene (full sequencing) or targets genetic variants more common in specific populations e.g. ancestry-based screening.
- The gene panel includes testing for the genetic condition suspicious in the family i.e. how comprehensive is the panel (BRCA1, BRCA2 only or additional genes listed in Table 2)
- If the results will be actionable. Not every gene offered on a gene panel necessarily has evidence-based guidelines to inform surveillance and can lead to uncertainty.
- Privacy of genetic information is ensured and how. Some companies leverage genetic and personal information allowing third-party access by entities such as research projects, advertising, law enforcement or pharmaceutical companies.
- The testing is performed within Canada or the sample is sent out of country where legislation around genetic testing, privacy etc. may be different.
It is also important to keep in mind that a negative result, although reassuring may not rule out the familial condition unless confirmation that the individual’s testing included analysis of a known familial variant. Additionally, a negative result does not mean the individual is not at increased risk for cancer. Risk assessment and screening recommendations are still based on family and personal health history. See more in What do the genetic test results mean?
What do the genetic test results mean?
For more information on genomic testing and results see our point of care tool. Most provincial genomic laboratories will have board-certified genetic counsellors who can answer your questions about genetic test results and are happy to take your calls.
With any suspected genetic condition, the best person in whom to begin genetic testing is always the person most likely to carry a pathogenic variant in a candidate gene. With hereditary cancer predispositions, this person is typically the one diagnosed with cancer at the youngest age (e.g. breast cancer at age 40), the one diagnosed with multiple cancers (e.g. breast and ovarian cancer, bilateral breast cancer), or the one diagnosed with a rare cancer (e.g. male breast cancer).
Affected individuals (have/had cancer diagnosis)
Positive result: A pathogenic/likely pathogenic (P/LP) variant in a cancer predisposition gene was identified. If the gene is associated with the person’s cancer diagnosis, this explains their cancer. Results may be used by oncology to inform treatment and/or surveillance plan and may allow for access to new effective therapies such as PARP inhibitors. The individual is likely at higher risk for subsequent cancer diagnoses. Additional screening and surveillance for other cancers may be indicated. First degree relatives (parents, siblings, offspring) have a 50% chance to inherit this gene change and to also be at risk. Relatives can have genetic testing for this familial variant.
Negative/uninformative result: No variants of clinical significance were identified in any of the genes on the cancer panel. Given the comprehensiveness of cancer gene panels, a negative result significantly reduces the likelihood of a hereditary cancer condition in the person tested. Depending on the family history, other relatives may still be eligible for genetic testing. Screening and surveillance recommendations for the individual and their relatives will be based on personal and family history, which may include high risk breast screening with MRI.
Variant of uncertain significance (VUS): A variant in a gene where the significance is not yet known. The laboratory cannot confidently determine if the gene variant identified is pathogenic or benign as available evidence is insufficient or conflicting. This result is reassuring as pathogenic variants in high/moderate risk cancer genes have been ruled out, however a hereditary cancer condition cannot be completely excluded. A board-certified genetic counsellor or a geneticist can help to interpret the laboratory report. No changes to medical management are indicated. Family members are typically not offered genetic testing for a VUS. Clinics and laboratories differ in their re-contact protocols, but generally a patient would be encouraged to re-contact their genetics provider in 2-5 years for updates on re-classification of the VUS they carry. Re-classification of a VUS could mean the variant is now determined to be pathogenic or benign.
Unaffected individuals (never had a cancer diagnosis)
Positive result: A P/LP variant in a cancer predisposition gene was identified. The individual is at increased risk for cancers associated with the gene (e.g. BRCA1 - breast and ovarian cancer, MLH1 - colon and endometrial). Screening and medical management recommendations can be made based on the combination of genetic test result and family history. First degree relatives (parents, siblings, offspring) have a 50% chance to inherit this gene change and to also be at risk. Relatives can have genetic testing for this familial variant.
Negative result:
True negative: The familial variant was not detected. This is where a pathogenic/ likely pathogenic variant in a cancer predisposition gene has already been identified in a relative. Testing was solely to detect the presence or absence of the familial pathogenic variant. This individual is not at increased risk for cancer. Depending on the family history, population-based cancer screening is still recommended.
Uninformative: No variants of clinical significance were identified in any of the genes on the cancer panel. Given the comprehensiveness of cancer gene panels, a negative result significantly reduces the likelihood of a hereditary cancer condition in the person tested, however without confirmation of a gene variant responsible for the family’s cancer, this is uninformative. Screening and surveillance recommendations for the individual and their relatives will be based on personal and family history, which may include high risk breast screening with MRI. Depending on the family history, other relatives may still be eligible for genetic testing.
Variant of uncertain significance (VUS): A variant in a gene where the significance is not yet known. The laboratory cannot confidently determine if the gene variant identified is pathogenic or benign as available evidence is insufficient or conflicting. A board-certified genetic counsellor or a geneticist can help to interpret the laboratory report. No changes to screening or medical management are indicated. Family members are typically not offered genetic testing for a VUS. Clinics and laboratories differ in their re-contact protocols, but generally a patient would be encouraged to re-contact their genetics provider in 2-5 years for updates on re-classification of the VUS they carry. Re-classification of a VUS could mean the variant is now determined to be pathogenic or benign.
A word about Variants of Uncertain Significance (VUS): A VUS is a result that leaves ambiguity for a patient and depending on a patient’s experience with cancer (personal and family), attitudes toward healthcare and education level, there may be an inappropriate expectation of increased monitoring based on the result.4 Discussions with healthcare practitioners are important in shaping a patient’s understanding of the result, managing uncertainty and setting expectations.4 It is also important to note that the rate of VUS is reported to be significantly higher in those of non-European ancestry (e.g. Hispanic, African, Asian and Pacific Islander). This has to do with the lack of diversity in clinical and research contexts and the under-representation of non-European groups in genomic databases that are used for interpretation by laboratories.5
Secondary Findings: As genetic technology expands, laboratories are increasing the number of genes included on hereditary cancer gene panels (tests that analyze multiple genes at the same time). As a result, testing may identify a pathogenic or likely pathogenic (P/LP) variant in a gene unrelated to the primary indication for testing. For example, genetic testing ordered because of suspicion for hereditary breast and ovarian cancer may identify a P/LP variant in a gene associated with hereditary colorectal cancer risk.
Supporting cascade genetic testing: Once a P/LP variant has been identified in a family, other relatives are eligible to have genetic testing for that variant and can have more accurate risk assessments, screening and management recommendations. Familial testing would require a copy of the genetic test report or a family letter that has the name of the familial genetic variant. Genetic counsellors can help facilitate communication of results among relatives by providing/sending family letters with de-identified information after ensuring appropriate consent to share genetic information is documented. Primary care practitioners can play a key role in cascade testing of hereditary conditions by discussing the benefits of sharing results, of genetic testing and by making referrals. A recent meta-analysis reported that about 1/3 of relatives were not informed about their risk to carry a P/LP variant and, of those that were informed, less than half went on to have genetic testing. Some barriers to disclosure of results to relatives include:6
- not being in close contact with relatives
- fear of causing relatives distress or anxiety, or finding the topic too distressing to bring up
- considering relatives to be either too old or too young to learn about the familial condition
- not understanding why sharing information is important
- feeling that genetic information is too personal to share
Both genetic and primary care clinicians are well positioned to provide support and education to overcome these barriers.
My patient had somatic testing. What does that mean? In cancer, somatic testing refers to genetic testing of the tumor. This is done to inform treatment, not carrier status. A pathogenic variant present in a tumor does not confirm a hereditary predisposition in the individual. If no pathogenic variant is detected, this does not rule out a hereditary predisposition in the individual and germline genetic testing may still be indicated.
Germline testing, typically performed on a blood sample, refers to genetic testing to identify inherited pathogenic variants - those that were present at conception in the egg or sperm.
- Somatic pathogenic variants are isolated to a tissue and are not passed on to offspring. These are acquired and sporadic.
- Germline pathogenic variants are expected to be present in every tissue. These are inherited and can be passed on to offspring.
What are the benefits and considerations of genetic testing?
Benefits:
- A positive result can explain why a person developed cancer and confirm a hereditary predisposition in the family
- Oncology may use the result to inform treatment
- Additional screening, surveillance and/or surgical recommendations may be suggested
- Clinical intervention for those with known hereditary cancer predispositions can improve outcomes7,8
- Relatives can have genetic testing for the familial variant
- At-risk relatives can be identified and other relatives without the familial variant can be reassured. Both can be given more accurate risk assessments3,9
- Positive health behaviours can be reinforced3,9
Considerations:
- Some may experience psychological distress over:
- the increased risk to develop cancer
- the possibility of passing an increased cancer risk to offspring
- the uncertainty of a negative result or a variant of uncertain significance
- the uncertainty of a cancer diagnosis, as not everyone with a genetic predisposition will develop cancer
- Family issues such as confidentiality concerns or estrangement may inhibit the transfer of information between relatives
- In Canada, the Genetic Non-Discrimination Act (GNDA) became law in 2017. This law protects Canadians and their genomic information. Some key points of the law are that GNDA:10
- Prohibits requiring someone to take a genetic test as a condition for a contract, service, or employment.
- Prohibits requiring disclosure of existing genetic test results to access insurance, employment, or services
- Makes it illegal to deny goods or services, or to change the terms of a contract (for example, higher premiums or refusal of insurance).
- Genetic test results cannot be used when making human resources decisions relating to employment
What are the cancer risks associated with an inherited predisposition?
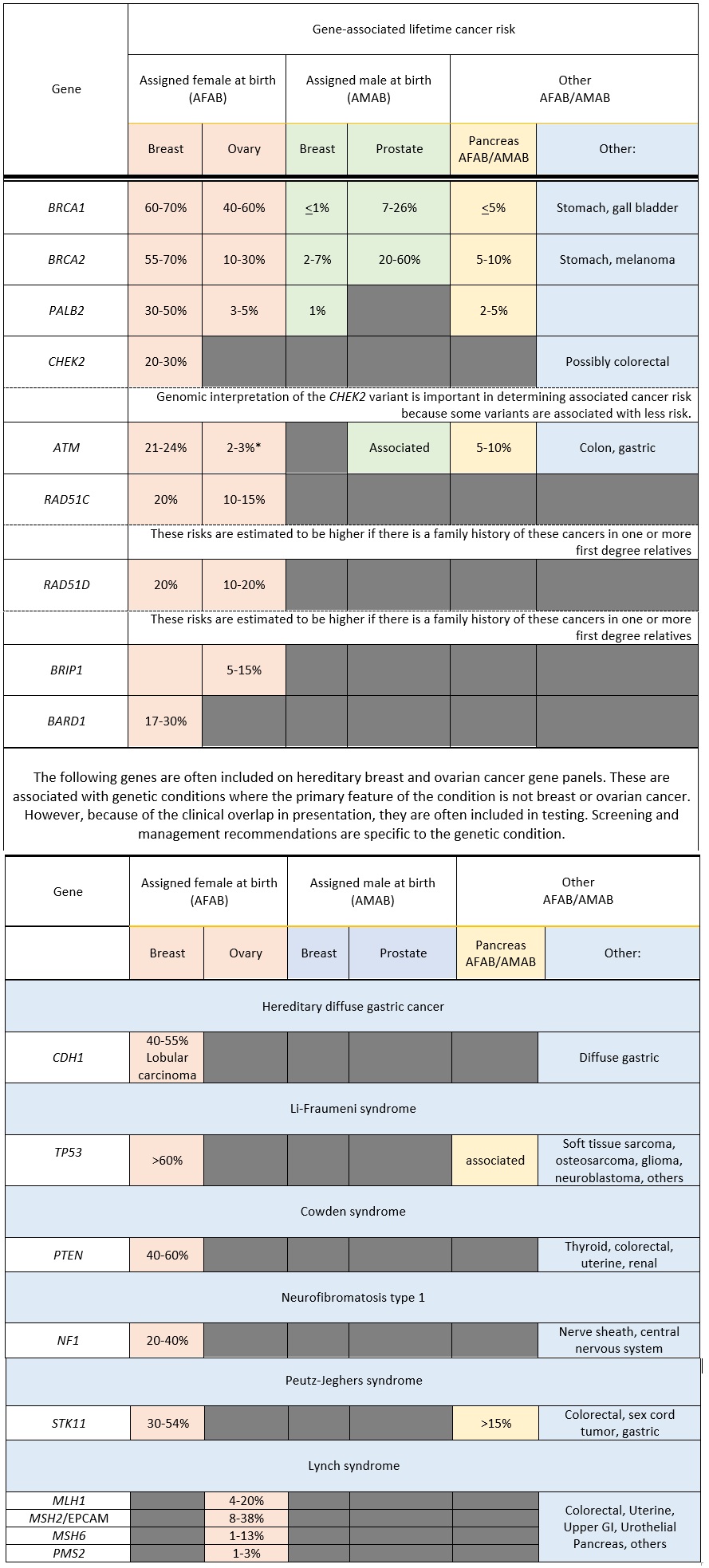
Many of the genes associated with an inherited predisposition to breast and ovarian cancer have a role in the cell’s ability to repair double-stranded DNA breaks (homologous repair), however associated cancers and lifetime risks vary between genes.11 Additionally an individual's personal risk may differ from published lifetime risks, depending on age, medical history and family history.3 Table 1 shows general population lifetime cancer risks. Table 2 shows approximate lifetime cancer risks associated with pathogenic/likely pathogenic (P/LP) variants in the indicated gene. Figures 2 and 3 show gene-associated lifetime breast and ovarian cancer risks as compared to the general population for those assigned female at birth.
Table 1. General population lifetime cancer risks where lifetime may be up to 70 or 80 years.12-14

Table 2. Approximate lifetime (70-80 years) cancer risks associated with P/LP variants in the gene listed. 1,3,12
Gene specific cancer surveillance and management recommendations
Recommendations for those with a pathogenic/likely pathogenic (P/LP) variant in CDH1, TP53, PTEN, NF1, STK11 or the Lynch syndrome genes are not covered in this resource. Screening, surveillance, and cancer risk reduction recommendations are gene-specific. For comprehensive overviews of these genetic predisposition conditions, see National Comprehensive Cancer Network, GeneReviews or contact your local genetics specialist.
A multidisciplinary approach involving medical, surgical, and genetic specialties is the best approach to managing those with a hereditary cancer predisposition.16 If your patient has not yet had genetic counselling to discuss their hereditary condition, consider referring so that appropriate specialists, perhaps those with special experience with hereditary cancer, can be identified and included in the care of your patient. If your patient had genetic counselling several years ago, consider reaching out to your local genetics specialist by phone or eConsult to inquire about any updated screening recommendations, as they evolve over time.
For those currently under the care of oncology, defer to oncology for screening and surveillance recommendations related to the current cancer diagnosis. Because those with hereditary cancer predispositions often have increased lifetime risk for additional cancers, management of those risks may be appropriate after treatment of the current cancer or may be combined with treatment for a current cancer (e.g. bilateral mastectomy versus unilateral for a BRCA1 P/LP variant carrier with a breast cancer diagnosis in one breast).
Screening
All screening should be offered with a discussion about the benefits, risks and limitations, such as early detection and reduced mortality, increased chance for false positive results and follow-up.
Breast: Assigned Female At Birth (AFAB)
Individuals with an increased lifetime risk for breast cancer should be familiar with their breasts and report changes to their healthcare clinician. For those with a high lifetime breast cancer risk, clinical breast exam (CBE) can be considered at 6-12 month intervals, particularly in the absence of the availability of MRI screening. Although there is no evidence to demonstrate that CBE reduces mortality, the rationale is the concern for interval breast cancers.1,3,17
High-risk breast cancer genes, BRCA1, BRCA2, PALB2, where lifetime risk is greater than 40%:
Increased screening for P/LP variant carriers results in earlier breast cancer diagnosis and improved outcomes.1 Recommendations may vary depending on a patient’s age, personal and family history, as well as access to screening modalities (e.g. MRI). In general, the recommendations for those AFAB who carry a P/LP variant in a high-risk breast cancer susceptibility gene are:1,3,18
- Annual screening with MRI and mammogram
- MRI starting at age 25-29 years
- Mammogram starting at age 30 years
- In the absence of availability of MRI, consider clinical breast exam every 6-12 months starting at age 25 years1,19
Moderate-risk breast cancer genes: ATM, BARD1, CHEK2, RAD51C, RAD51D, where lifetime risk is 40% or less, but greater than average:
As with high-risk breast cancer susceptibility genes, screening recommendations may vary depending on a patient’s age, personal and family history, as well as access to screening modalities (e.g. MRI). In general, the recommendations for those AFAB with a P/LP variant in a moderate-risk breast cancer susceptibility gene are:1,3
- Annual mammogram
- Starting at age 40 years
- Consider annual breast MRI
- Starting at age 40 years (30–35 years for the ATM gene)
- Factors to consider: lifetime breast cancer risk based on validated risk model (e.g CanRisk), age, breast density, family history, gene involved
Breast: Assigned Male At Birth (AMAB)
For those AMAB who carry a P/LP variant in the BRCA1, BRCA2 or PALB2 gene, the recommendations are:3
- Breast self-exam training and education
- Starting at age 35 years
- Annual clinical breast exam
- Starting at age 35 years
For those AMAB who carry a P/LP variant in the BRCA2 gene, consider:
- Annual mammogram or ultrasound screening
- Starting at age 50 or 10 years before the youngest male breast cancer diagnosis in the family, whichever comes first3
Ovary: relevant to those with a P/LP variant in the BRCA1, BRCA2, RAD51C, RAD51D, BRIP1, ATM, PALB21,3,17
Screening for ovarian cancer (e.g. transvaginal ultrasound with serum CA-125) is not routinely recommended. Studies assessing sensitivity and specificity of modalities and whether these improve survival have yielded mixed results.
Those with ovaries are encouraged to be aware of symptoms of ovarian cancer and to promptly report any to their healthcare clinician. Discussion of surgical management for cancer risk reductions should be offered to those with a high lifetime risk (>10%) for ovarian cancer.
Prostate: relevant to those with a P/LP variant in the BRCA2, and possibly BRCA1 and ATM1,3,20
Those AMAB with a P/LP variant in the BRCA2 gene are at significantly increased risk for prostate cancer as well as onset at younger age and more aggressive disease. Screening recommendations for these individuals are:
- Annual serum prostate specific antigen (PSA)
At this time there is no strong evidence to support prostate cancer screening in those AMAB who carry a BRCA1 or ATM P/LP variant. The Canadian Urological Association provides recommendations and guidance for prostate cancer screening for those at average and increased risk (e.g. family history) for prostate cancer.20
Pancreas: relevant to those with a P/LP variant in BRCA2 and ATM, BRCA1 and PALB2 1,3,21
Pancreatic cancer screening may not routinely be recommended unless there is a family history. Depending on availability of screening, individuals with BRCA2 or ATM P/LP variants may be offered pancreatic cancer screening in the absence of a family history.
Generally, an individual with at least one affected first or second degree relative may be considered eligible for pancreatic cancer screening. Screening may vary by region, access to screening modality and/or access to research setting and appropriate expertise. In general, screening recommendations are:
- Annual contrast-enhanced MRI and/or endoscopic ultrasound
- Starting at age 50 years or 10 years younger than the youngest diagnosis of pancreatic cancer in the family
Cancer risk reduction
SURGICAL MANAGEMENT
Breast: Assigned Female At Birth (AFAB)
High-risk breast cancer genes, BRCA1, BRCA2, PALB2:1,3,18
For those with a P/LP variant in BRCA1 or BRCA2 gene, bilateral risk-reducing mastectomy (BRRM) provides a high degree of protection against cancer. This decision is very personal and should include multidisciplinary consultations to consider various factors such as: risks, reconstruction options, degree of protection, psychosocial aspects (decreased cancer worry, impact on body image), the patient’s age and life expectancy, and family history. The benefits of RRM are likely greatest if carried out from the age of 30. Beyond age 55 the evidence for benefit of RRM is weak.
Because of the increased risk to develop a contralateral breast cancer some individuals may consider mastectomy versus lumpectomy and RRM of the other breast at the time of treatment for a breast cancer.
Those with a P/LP variant in the PALB2 gene may also benefit from discussion of BRRM with a multidisciplinary team to consider risks and benefits.
Moderate-risk breast cancer genes: ATM, BARD1, CHEK2, RAD51C, RAD51D, where lifetime risk is 40% or less, but greater than average:1,3,17
Evidence is insufficient to recommend BRRM based on genetic test results alone, however with individualised risk assessment, including family history, and careful consideration with multidisciplinary consultation, RRM may be considered on a case-by-case basis.
Surgery significantly reduces but does not eliminate the risk of cancer as some breast tissue may remain.
Ovary: relevant to those with a P/LP variant in the BRCA1, BRCA2, PALB2, BRIP1, RAD51C, RAD51D 1,3,17,18
In the absence of reliable screening methods for early detection of ovarian cancer, risk-reducing salpingo-oophorectomy (RRSO) is a reasonable option for those at high risk for ovarian cancer. RRSO may be considered when the risk of developing ovarian cancer exceeds that of the average population risk (~1%), however historically 10% or greater is generally accepted as high risk. Where the lifetime ovarian cancer risk is <10% but higher than average, individualized risk assessment including genetic test result, age, and family history should be considered in decision making.
The decision to undergo RRSO is complex and should not be made lightly, given the impact of premature menopause and related impact on quality of life. Ideally the decision is made in consultation with a gynecologic oncologist.
For those with a P/LP variant in the gene listed, RRSO recommendations, taking into consideration family history, are:
- BRCA1 gene: age 35-40 years
- BRCA2 gene: age 40-45 years: can be delayed as onset tends to be later than BRCA1
- BRIP1, RAD51C, RAD51D and PALB2 genes: age 45-50 years
- ATM gene: there is insufficient evidence for RRSO based on genetic test results alone and management recommendations should be made based on family history
Family history should always be considered when deciding about most appropriate age for RRSO as an earlier age may be more appropriate. Surgery significantly reduces but does not eliminate the risk of cancer as some ovarian tissue may remain.
MEDICATION OPTIONS1,3
Those assigned female at birth may benefit from discussion with a high-risk clinician about the benefits and limitations of chemoprevention for cancer risk reduction.
Breast:
Chemoprevention may be an option for women who postpone, or do not undergo, RRM, those with a contralateral breast cancer risk, or those who have an increased lifetime breast cancer risk not high enough to warrant surgery (e.g. some pathogenic variants in CHEK2). For post-menopausal women with an elevated lifetime risk of breast cancer, selective estrogen receptor modulators [tamoxifen and raloxifene], and aromatase inhibitors [anastrozole and exemestane] have been shown to reduce breast cancer incidence by ~30%- 60%. Data specifically in those with BRCA1 or BRCA2 P/LP variants is limited.
Ovary:
Use of the oral contraceptive pill (OCP) is associated with 40%-60% lower risk for ovarian cancer in those who carry a P/LP variant in BRCA1 or BRCA2. Data is conflicting as to the associated increased risk for breast cancer in these individuals with OCP use. Discussion with a multidisciplinary team to weigh the benefits, risks and consider family and personal history is recommended.
Reproductive considerations (family planning)
For certain hereditary cancer genes, if an individual and their reproductive partner are both carriers of a P/LP variant in the same gene, they would have a 25% chance to have a child affected with a rare autosomal recessive condition.
Carrier testing for the partner of a P/LP variant in a hereditary cancer gene is not routinely offered. In circumstances of a significant family history suggestive of hereditary breast and ovarian cancer or a consanguineous union (where both partners are related by blood, such as first cousins or closer) genetic counselling and consideration of carrier testing could be offered.
An exception is the ATM gene, where the carrier frequency is 1 in 100. Reproductive partners could be offered carrier testing for the ATM gene to assess reproductive risk.
Fanconi anemia
If an individual and their reproductive partner are both carriers of a P/LP variant in the BRCA1, BRCA2, PALB2, BRIP1, or RAD51C gene, they would have a 25% chance to have a child affected with a rare autosomal recessive condition called Fanconi anemia (FA), in addition to an increased risk for pregnancy loss. FA is a rare inherited DNA repair disorder. It is the most common cause of inherited bone marrow failure. FA is also associated with congenital anomalies and cancer predisposition. Both members of the couple must be carriers of P/LP variants in the same gene in order to be at risk to have a child affected with FA.
Ataxia telangiectasia
If an individual and their reproductive partner are both carriers of a P/LP variant in the ATM gene, they would have a 25% chance to have a child affected an autosomal recessive condition known as ataxia-telangiectasia (AT). AT is a progressive neurological disorder that also affects the immune and other body systems and is characterized by progressive difficulty with coordinating movements (ataxia) beginning in early childhood, usually before age 5. Small clusters of enlarged blood vessels called telangiectasias, which occur in the eyes and on the surface of the skin, are also characteristic of this condition.
The carrier frequency of P/LP variants in the ATM gene is about 1 in 100. If an individual of reproductive age has one variant in the ATM gene, their reproductive partner could have genetic testing to see if they are at risk of having a child with AT.
Carriers of one P/LP variant in the ATM gene will not develop AT.
Constitutional Mismatch Repair Deficiency
If an individual and their reproductive partner are both carriers of a P/LP variant in the same Lynch syndrome gene, they would have a 25% chance to have a child affected with a rare autosomal recessive condition called constitutional mismatch repair deficiency (CMMRD). CMMRD is associated with a very high cancer risk in childhood.
Genetic testing of minors
Guidelines recommend that unless there is evidence for timely medical benefit or actionable management in the pediatric period, genetic testing for adult-onset conditions should be deferred until the child/minor is capable of deciding whether to be tested. Clinicians are not obligated to comply with requests from parents to test healthy children. There may be exceptions such as where the wait for testing itself may cause an undue psychological burden for the child. Requests for testing by competent, well-informed adolescents may be considered in conjunction with comprehensive genetic counselling.22,23
References
- Sessa C, Balmaña J, Bober SL, et al; ESMO Guidelines Committee. Risk reduction and screening of cancer in hereditary breast-ovarian cancer syndromes: ESMO Clinical Practice Guideline. Ann Oncol. 2023 Jan;34(1):33-47. PMID: 36307055
- Ashton-Prolla P, Giacomazzi J, Schmidt AV, et al. Development and validation of a simple questionnaire for the identification of hereditary breast cancer in primary care. BMC Cancer 2009; 9:283 Licence: http://creativecommons.org/licenses/by/2.0/
- National Comprehensive Cancer Network (NCCN). NCCN Clinical Practice Guidelines in Oncology. Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic Version 3.2026.Feb 19 2026. National Comprehensive Cancer Network. Available from: https://www.nccn.org/professionals/physician_gls/pdf/genetics_bop.pdf [Accessed May 2026]
- Mighton C, Clausen M, Shickh S, Baxter NN, Scheer A, Sebastian A, Muir SM, Kim THM, Glogowski E, Schrader KA, Regier DA, Kim RH, Lerner-Ellis J, Bayoumi AM, Thorpe KE, Bombard Y. How do members of the public expect to use variants of uncertain significance in their health care? A population-based survey. Genet Med. 2023 May;25(5):100819. PMID: 36919843.
- Ndugga-Kabuye MK, Issaka RB. Inequities in multi-gene hereditary cancer testing: lower diagnostic yield and higher VUS rate in individuals who identify as Hispanic, African or Asian and Pacific Islander as compared to European. Fam Cancer. 2019 Oct;18(4):465-469. PMID: 31531760
- Ahsan MD, Levi SR, Webster EM, et al. Do people with hereditary cancer syndromes inform their at-risk relatives? A systematic review and meta-analysis. PEC Innov. 2023 Feb 17;2:100138. PMID: 37214514
- Moran A, O’Hara C, Khan S, et al. Risk of cancer other than breast or ovarian in individuals with BRCA1 and BRCA2 mutations. Fam Cancer 2012; 11(2):235-42
- Narod SA, Offit K. Prevention and management of hereditary breast cancer. J Clin Oncol 2005; 23(8):1656-63
- Horsman D, Wilson BJ, Avard D, et al. Clinical management recommendations for surveillance and risk-reduction strategies for hereditary breast and ovarian cancer among individuals carrying a deleterious BRCA1 or BRCA2 mutation. J Obstet Gynaecol Can 2007; 29(1):45-60.
- Cowan JS, Kagedan BL, Graham GE, et al. Health care implications of the Genetic Non-Discrimination Act: Protection for Canadians' genetic information. Can Fam Physician. 2022;68(9):643-646
- Murfuni, I.; Rass, U. Chapter 8—Targeting homologous recombination repair in cancer. In DNA Repair in Cancer Therapy, 2nd ed.; Kelley, M.R., Fishel, M.L., Eds.; Academic Press: Cambridge, MA, USA, 2016; pp. 225–275. [Google Scholar]
- Petrucelli N, Daly MB, Pal T. BRCA1- and BRCA2-Associated Hereditary Breast and Ovarian Cancer. 1998 Sep 4 [Updated 2026 Mar 25]. In: Adam MP, Mirzaa GM, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2023. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1247/
- Canadian Cancer Statistics Advisory in collaboration with the Canadian Cancer Society, Statistics Canada and the Public Health Agency of Canada. Canadian Cancer Statistics: A 2022 special report on cancer prevalence. Toronto, ON: Canadian Cancer Society; 2022. Available at: cancer.ca/Canadian-Cancer-Statistics-2022-EN (accessed [30 Aug 2023]).
- Surveillance, Epidemiology, and End Results (SEER) Program (www.seer.cancer.gov) SEER*Stat Database: Incidence - SEER Research Data, 8 Registries, Nov 2021 Sub (1975-2020) - Linked To County Attributes - Time Dependent (1990-2020) Income/Rurality, 1969-2020 Counties, National Cancer Institute, DCCPS, Surveillance Research Program, released April 2023, based on the November 2022 submission
- Li S, Silvestri V, Leslie G, et al. Cancer Risks Associated With BRCA1 and BRCA2 Pathogenic Variants. J Clin Oncol. 2022 May 10;40(14):1529-1541. PMID: 35077220
- Samadder NJ, Giridhar KV, Baffy N, et al. Hereditary Cancer Syndromes-A Primer on Diagnosis and Management: Part 1: Breast-Ovarian Cancer Syndromes. Mayo Clin Proc. 2019 Jun;94(6):1084-1098. PMID: 31171119.
- Hanson H, Kulkarni A, Loong L, et al. UK consensus recommendations for clinical management of cancer risk for women with germline pathogenic variants in cancer predisposition genes: RAD51C, RAD51D, BRIP1and PALB2. J Med Genet. 2023 May;60(5):417-429. PMID: 36411032
- Tischkowitz M, Balmaña J, Foulkes WD, et al. Management of individuals with germline variants in PALB2: a clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2021 Aug;23(8):1416-1423. PMID: 33976419.
- Hettipathirana T, Macdonald C, Xie J, et al. The value of clinical breast examination in a breast cancer surveillance program for women with germline BRCA1 or BRCA2 Med J Aust. 2021 Nov 15;215(10):460-464. PMID: 34420218.
- Mason RJ, Marzoik K, Finelli A, et al. UPDATE – 2022 Canadian Urological Association recommendations on prostate cancer screening and early diagnosis: Endorsement of the 2021 Cancer Care Ontario guidelines on prostate multiparametric magnetic resonance imaging. Can Urol Assoc J 2022;16(4):E184-96. http://dx.doi.org/10.5489/cuaj.7851
- Goggins M, Overbeek KA, Brand R, et al. Management of patients with increased risk for familial pancreatic cancer: updated recommendations from the International Cancer of the Pancreas Screening (CAPS) Consortium. Gut. 2020 Jan;69(1):7-17. Epub 2019 Oct 31. Erratum in: Gut. 2020 Jun;69(6):e3. PMID: 31672839
- Moore AM, Richer J. Genetic testing and screening in children. Paediatr Child Health. 2022 Jul 18;27(4):243-253. PMID: 35859684
- COMMITTEE ON BIOETHICS; COMMITTEE ON GENETICS, AND; AMERICAN COLLEGE OF MEDICAL GENETICS AND; GENOMICS SOCIAL; ETHICAL; LEGAL ISSUES COMMITTEE. Ethical and policy issues in genetic testing and screening of children. Pediatrics. 2013 Mar;131(3):620-2. PMID: 23428972.
Authors: S Morrison MS CGC, JE Allanson MD FRCPC, S Walji MD CCFP and JC Carroll MD CCFP

