![]()

(July 2026)
Download the whole PDF here. Check out our FH point of care tools or our more concise summary, GECKO on the run.
| Bottom line: Familial hypercholesterolemia (FH) is a common (~1/250) autosomal dominant condition that results in a 6- to 22-fold increase in premature cardiovascular disease (CVD) and death. Early diagnosis and treatment can normalize life expectancy. Key features of FH are elevated LDL-C ≥5 mmol/L, early onset CVD (<55 years in men, <65 years in women), cholesterol deposition in the tendons (xanthomata) and/or around the eyes (xanthelasma), arcus cornealis with onset <45 years, and family history of early onset CVD or hyperlipidemia requiring treatment. In Canada, a diagnosis of FH is typically based on an individual’s clinical presentation and history as outlined in the Canadian Cardiovascular Society algorithm. Genetic testing is not widely clinically available in Canada with some exceptions. A clinical diagnosis guides treatment and screening of family members. Once a person is diagnosed with FH, cascade screening of family members using measurement of LDL-C levels and/or genetic testing is recommended. This enables early identification and treatment of at-risk individuals, with statins as first-line treatment. |
Jump to a section by clicking a link below:
- What is Familial Hypercholesterolemia?
- How is familial hypercholesterolemia diagnosed?
- How to order genetic testing for familial hypercholesterolemia?
- Cascade screening for family members
- What do the genetic test results mean?
- What are the benefits and considerations of genetic testing for familial hypercholesterolemia?
- Surveillance and Management
- Resources
- References
- Additional readings
What is Familial Hypercholesterolemia?
Familial hypercholesterolemia (FH) is an autosomal dominant genetic condition where the uptake of low-density lipoprotein cholesterol (LDL-C) into cells is either decreased or inhibited. This results in lifetime exposure to very high levels of LDL-C. FH is the most common genetic disorder causing premature cardiovascular disease (CVD) and death in both men and women. FH is both underdiagnosed and undertreated worldwide despite the knowledge that early diagnosis and treatment can normalize life expectancy.1-3 It is estimated that roughly 1 in 250 Canadians has FH, and that only about 10% have been identified.1 The global prevalence of FH in the general population is about 1 in 300 and is 10- to 20-fold more common among individuals with ischemic heart disease, particularly premature ischemic heart disease.4
What do I need to know about the genetics of familial hypercholesterolemia?
Most cases (up to 80%) of familial hypercholesterolemia (FH) are caused by pathogenic/likely pathogenic (P/LP) variants (what used to be called mutations) in the LDL receptor gene LDLR, in which > 3000 different P/LP variants have been identified.2,5,6 The LDLR protein binds LDL, which is the major cholesterol-carrying lipoprotein of plasma, and transports LDL into cells by endocytosis. P/LP variants in the LDLR gene can reduce the number of LDL receptors produced within cells or disrupt the ability of the receptor to bind LDL particles.2 P/LP variants in APOB disrupt binding of LDL particles to the receptor, while P/LP variants in PCSK9 cause increased degradation of the receptor. These mechanisms lead to elevated LDL-C levels and premature development of atherosclerotic plaque.
Additional genes (e.g. ABCG5, ABCG8, APOE, LDLRAP1, LIPA) are known to be associated with FH, although very rare and atypical. With advances in genetic testing technology, additional rare genes can be added to gene panels with little extra cost. Genetic testing for FH may involve a gene panel with comprehensive analysis of three or more genes or may be targeted ancestry-based testing looking for the presence or absence of specific P/LP variants.7
Pattern of inheritance
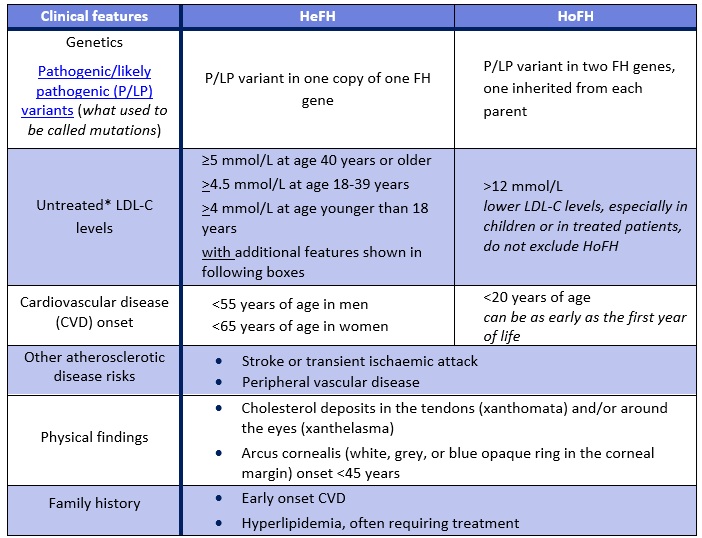
FH is typically inherited in an autosomal dominant manner. FH can be present in a heterozygous form (HeFH), where only one copy of a FH-causing gene contains a P/LP variant. FH can also be present in a homozygous form (HoFH) where an individual has a P/LP variant in both copies of a FH-causing gene. The two P/LP variants can be identical or different. Rarely there is a P/LP variant in one copy of two different FH genes (digenic inheritance). All individuals with HoFH have an extremely high risk of early onset cardiovascular disease.1,3 If both parents have HeFH, their child has a 25% chance to have HoFH, which is associated with an extremely high CVD risk.
Table 1. Clinical features of familial hypercholesterolemia in heterozygotes (HeFH) and homozygotes (HoFH).*The CardioRisk app has a validated algorithm to impute a baseline value from LDL-C levels while on lipid lowering medications, additionally it can be used for the clinical diagnosis of FH, assessing the degree of severity of FH for new patients and helps facilitate FH diagnosis.
How common is familial hypercholesterolemia?
About 1 in 250 Canadians is thought to have heterozygous familial hypercholesterolemia (HeFH), however FH is significantly under-recognized in Canada.1 Homozygous-FH (HoFH) is much rarer, and more severe and is expected to affect between 1 in 250,000 and 1 in 1,000,000 Canadians.8 FH is more common in certain populations due to founder effects: in certain areas of Quebec, the prevalence is as high as 1 in 80.9
How is familial hypercholesterolemia diagnosed?
The Canadian Cardiovascular Society (CCS) recommends the use of the Canadian diagnostic criteria for FH proposed by the Familial Hypercholesterolemia Canada (FHCanada) network (Figure 1).10 While these criteria are relatively new, they are less complicated than those published by the Dutch Lipid Clinic Network (DLCNC) (Table 2) or the Simon Broome Registry (Table 3) and have been validated against each of these criteria, which are internationally accepted for the diagnosis of HeFH.10 The Simon Broome Registry criteria include lower thresholds for children with suspected FH.11 Neither the DLCNC nor Simon Broome Registry criteria were designed to diagnose HoFH, for which other criteria have been suggested.8 The European Atherosclerosis Society has recently published clinical and genetic diagnostic criteria for HoFH.8 Genetic testing is not necessary for diagnosis and is not yet routinely clinically available in most of Canada. See the How to order the genetic testing for FH for more.
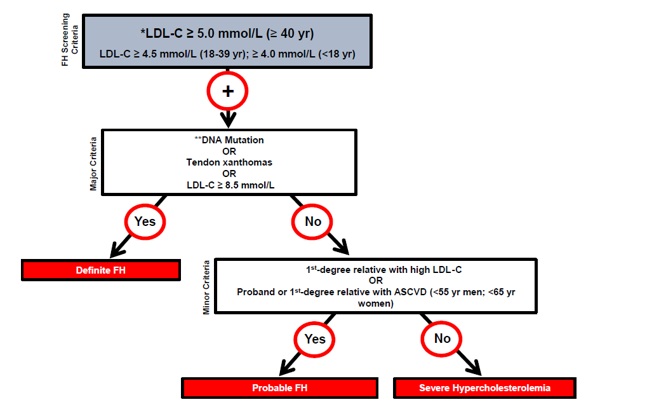
Figure 1. Canadian criteria for the clinical diagnosis of familial hypercholesterolemia (FH). From Ruel I et al, 201810. Reprinted with permission under the CC BY-NC-ND license https://creativecommons.org/licenses/by-nc-nd/4.0/. DOI: 10.1016/j.cjca.2018.05.015
ASCVD: atherosclerotic cardiovascular disease; LDL-C: low-density lipoprotein cholesterol. * Secondary causes of high LDL-C should be ruled out (severe or untreated hypothyroidism, nephrotic syndrome, hepatic disease [biliary cirrhosis], medication, especially antiretroviral agents) ** DNA mutation refers to the presence of a known FH-causing variant in a FH gene in the individual or a first-degree relative. FH diagnosis in a patient with a P/LP variant but normal LDL-C levels is unclear. Yearly follow-up of the individual is suggested, and cascade screening of family members should be initiated.
 |
The LDL-C levels cited are untreated levels. For a patient who is on lipid lowering medications, the CardioRisk CalculatorTM app has a validated algorithm to assign a baseline value. |
Table 2. Dutch Lipid Clinic Network Criteria for Diagnosis of FH.3
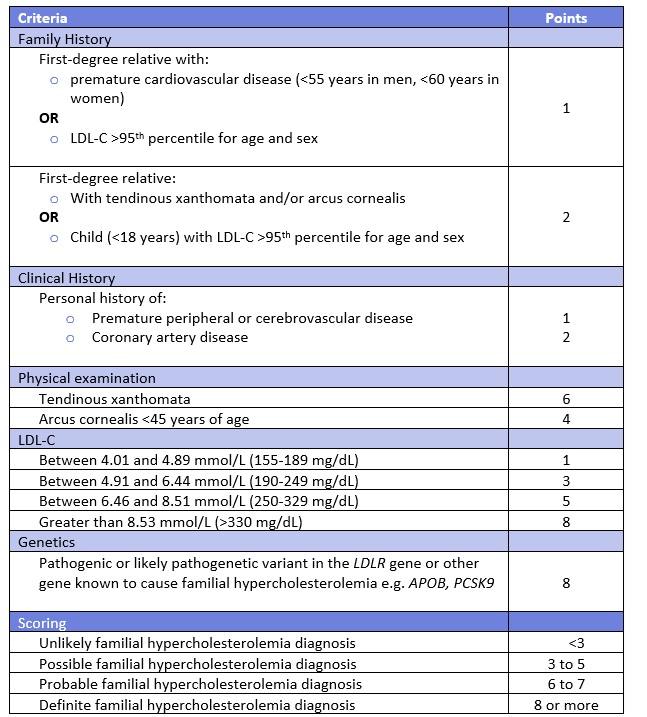
Table 3. Simon Broome Registry Criteria for Diagnosis of FH. 11,15

How to order genetic testing for familial hypercholesterolemia?
While genetic testing is not necessary for diagnosis it can confirm a diagnosis of FH, identify at-risk relatives early through cascade testing, refine cardiovascular risk assessment, and has shown to improve treatment uptake, adherence, and LDL cholesterol reduction.23
Genetic testing for FH is not available throughout Canada and some provinces have ordering restrictions.23 Contact your local genetics centre for support. See below for how genetic testing can be accessed in your province.
- Testing must be ordered by geneticist in Manitoba and Newfoundland and Labrador.
- In addition to geneticists, specialist physicians in endocrinology, cardiology and internal medicine can order genetic testing for FH in Alberta, Saskatchewan and Nova Scotia.
- Genetic testing for FH is not yet clinically available in British Columbia.
- In Ontario and Quebec, genetic testing can be ordered by any physician and does not require a referral for a specialist assessment
Genetic testing in Québec, select the laboratory closest to you: (July 2026)
- The Core Molecular Diagnostic Laboratory at the McGill University Health Centre: Core Molecular Diagnostics Requisition (DM 5891)
- CHU Sainte Justine Molecular Laboratory
Genetic testing in Ontario, select the laboratory closest to you: (July 2026, alternately you can use the Ontario Genetic Test Directory to search):
- London Health Sciences Centre (LHSC Requisition) molecular laboratory
- Trillium Health Partners – Credit Valley Site (THP Requisition)
- Hamilton Regional Laboratory Medicine Program (Requisition)
Cascade screening for family members
The most cost-effective approach for identification of new FH cases is cascade screening of family members of the first individual with a confirmed diagnosis, known as the index case.4,11,13 Data from the UK have shown that cascade screening reduces the average age at which an individual is diagnosed and results in an increased number of individuals who are treated with statins and have subsequent lowered lipid levels.14
The Canadian Cardiovascular Society (CCS) recommends screening of first-degree relatives of the index case.1 Screening can include lipid profiles of relatives and/or genetic testing for a known familial P/LP variant, when available. Each newly diagnosed individual becomes a new index case and cascade screening of relatives continues.
When using a genetic testing approach, testing relatives for a known familial P/LP variant, positive results will identify at-risk relatives and negative results would reassure those at population risk. When ordering genetic testing for relatives it is important to include documentation of the familial genetic variant either with a molecular report or a family letter. This ensures accurate interpretation of testing.
What do the genetic test results mean?
Test results fall into three categories:
Positive: causative pathogenic variant detected in an FH gene
- This confirms a genetic etiology and a diagnosis of FH.
- These results can be used to guide management.
- Genetic testing can be offered to relatives for the familial gene variant.
Negative
Uninformative: no causative pathogenic variant detected in any FH gene tested
- A diagnosis of FH is neither confirmed nor ruled out.
- If testing was limited to ancestry-based screening (where only select genetic variants in select genes were analysed), expanded testing may be considered, if available.
- Genetic testing cannot be offered to unaffected relatives. Lipid screening can be used for at-risk relatives.
- Re-referring to genetics can be considered if/when genetic testing improves.
True negative: the familial P/LP variant is not detected
- This is where genetic testing is offered to an unaffected relative after the causative pathogenic gene variant has been identified.
- The individual does not have the familial condition and is not at increased risk for dyslipidemia.
- This individual’s offspring would also not have increased risk for dyslipidemia.
Variant of unknown significance: variant in FH-related gene is detected, but there is insufficient evidence to determine if it is truly associated with disease
- A diagnosis of FH is neither confirmed nor ruled out.
- Genetic testing is not offered to unaffected relatives. Lipid screening can be used for at-risk relatives.
- Segregation studies may be considered. This is where genetic testing is offered to other affected relatives to try and track if the VUS is present in others with the condition.
- Re-referring to genetics at a later date (3-5 years) can be considered as over time a VUS may be reclassified as benign or pathogenic.
Visit the GECKO site to find more resources on genetic test results including a point of care tool.
What are the benefits and considerations of genetic testing for familial hypercholesterolemia?
Benefits
Confirmation of diagnosis:15 Genetic testing is a key approach to the diagnosis of definite FH. While clinical criteria can be used (Table 2 and 3) for diagnosis, there are limitations to using the classical presentation as a criterion since few affected persons will exhibit physical findings (e.g. xanthomas, xanthelasmas) at the time of testing. Additionally, there are limitations to use of family history of cardiovascular disease in FH diagnosis. Hypercholesterolemia or heart disease could be masked in relatives who are receiving lipid lowering treatment. Also, the penetrance of FH is very strong but not complete (i.e. not all variant carriers will develop hypercholesterolemia), and self-reported family history is not always accurate. Screening for FH based on family history alone has shown to miss 30-60% of cases.16
Refining risk stratification:15 Detection of a pathogenic variant in an FH gene indicates higher cardiovascular risk (compared to those at the same LDL-C levels) and the need for more aggressive LDL-C reduction. The use of conventional cardiovascular risk calculators in individuals with FH is not recommended as these greatly underestimate lifetime CVD risk.1,2, 17
Cascade testing: Once a genetic diagnosis has been established in an individual, their relatives can seek genetic testing to look for the presence or absence of the same genetic variant. A negative result would mean the individual is at population risk for CVD (unless there are additional risk factors17. A positive result would confirm a diagnosis of FH and appropriate early screening and interventions can be initiated.
Value to children and adolescence:15 Statin treatment in children identified as carrying a pathogenic variant in an FH gene may begin as young as 8 years of age. Children with FH who start a statin have statistically lower event rates than their affected parents. Left untreated, children with FH will be at higher risk of coronary events as adults because of the cumulative burden of elevated LDL-C levels.
Considerations
Sensitivity: Current genetic testing does not detect all possible genetic causes of FH and so a negative test result would not rule out an FH diagnosis.
Accessibility: Genetic testing for FH is not equitably available across Canada. Some provinces (Ontario, Quebec) offer testing through provincial laboratories. Other locations do not have in-province access. It may be possible to request funding for out-of-country testing or through research avenues.
Surveillance and Management
Adults
For more on the management and screening of dyslipidemia in adults, please see the 2021 guidelines from the Canadian Cardiovascular Society (CCS).17 These recommendations include universal lipid screening of men and women at average risk age 40 years and older, or earlier screening for those at increased risk for CVD. CCS also recommends regular atherosclerotic cardiovascular disease (ASCVD) risk assessments every 5 years for men and women aged 40-75 years, using a validated risk model (e.g. Framingham Risk Score [FRS] or the Cardiovascular Life Expectancy Model [CLEM]). Recent PEER simplified guidelines18 suggest for those at average risk of CVD, universal lipid screening at starting age 40 for those assigned male at birth and at age 50 for those assigned female at birth. If CVD risk factors are present, screening can be considered younger.
For management and screening recommendations for those individuals with FH, see the 2018 CCS Position Statement and the more recently published evidence-based guidelines by The International Atherosclerosis Society19 and the recent review article Stein JAMA 2026 Epub ahead of print. PMID: 42371647.
The use of conventional cardiovascular risk calculators in individuals with FH is not recommended as these greatly underestimate lifetime CVD risk.1,2,19 FH-specific cardiovascular risk calculators (e.g. the FH Risk Score20, SAFEHEART 19) should be considered to assess the risk of ASCVD in those with FH. Routine assessment and stratification of the risk of ASCVD in all patients with FH should be used to guide personalized treatment and management.19 Referral to specialist for risk stratification can be considered.
Those with homozygous status (HoFH, two pathogenic/likely pathogenic (P/LP) variants in an FH gene) should be referred to a specialized lipid centre.1
A genetic diagnosis of FH in a person with a P/LP variant but normal LDL-C levels is unclear. Yearly follow-up of the individual is suggested, and cascade screening of family members should be initiated.1
Medications
Statins are the drug class of choice for individuals with one P/LP variant in a FH gene (HeFH). Observational studies have shown a dramatic decrease in cardiac events in statin-treated individuals with FH.1 LDL-C should be lowered as fast and as far as possible.3 The CCS recommends a >50% reduction of LDL-C as primary prevention and an ideal goal of LDL-C <1.8 mmol/L for secondary prevention.17 Non-fasting lipid profiles should be used to monitor treatment in those whose treatment is stable.20 The use of high-dose statins alone is usually sufficient to achieve LDL-C reduction; however, some individuals with FH will require combination (e.g. ezetimibe) and/or emerging therapy (e.g. PCKS9 inhibitor) to obtain optimal LDL-C. Specialist referral is recommended for these cases.1-3,17,23
For the most recent recommendations on management and treatment of individuals with HoFH please see Cuchel et al. 2023.8
Lifestyle
All families with FH (including children and adolescents) should be counselled about the importance of lifestyle modification and heart healthy behaviour1-3,17,19 such as:
- Smoking cessation and avoidance of passive smoking
- Diet
- High in fibre (soluble), plant sterols/stanols and unsaturated fatty acids
- Low in trans and saturated fatty acids, refined sugars
- Exercise
- Daily activity beginning early in life
- Maintenance of ideal body weight
- Stress reduction
Pregnancy
Statins have been considered a teratogen on the basis of animal studies, however human studies have shown conflicting results.20 For most persons assigned female at birth who are of reproductive age, an effective birth control method is recommended with discontinuation of statin therapy ideally 3 months prior to planned pregnancy or at the time of a positive pregnancy test.1,20,21 Bile acid sequestrants can be considered to treat hypercholesterolemia, ideally 3 months before a planned pregnancy, as well as during pregnancy and lactation.19 The CCS recommends referral to a maternal-fetal-medicine or obstetrical specialist for management of lipid reducing therapies.17 A pregnant person with FH and additional risk factors, e.g. established ASCVD, should be referred to a speciality lipid clinic for further treatment advice.
Children/Adolescents
There remains controversy as to whether screening in childhood for FH should be implemented.1 A recent systematic review by the US Preventive Services Task Force found that the use of statins in children to reduce lipid levels appears beneficial, and observational studies show there is long-term benefit.20 The Canadian Pediatric Society22 recommend universal lipid screening (fasting or non-fasting, non-HDL-C or LDL-C) be performed after 2 years of age within the first decade of life.21,22 Selective screening at anytime can be considered when there is a positive family history of premature CVD or dyslipidemia, or other cardiovascular risk factors.21,22 Reverse cascade screening of parents is recommended when a child is found to have FH.1,21,22
The ideal age to begin treatment is between ages 8-12 years based on current randomized control trials.21 Pharmacological treatment can be considered, incorporating clinical judgement, family and patient preferences,1,19,21,22 at:
- Age 8-10 years when LDL-C concentration >4.9 mmol/l, recorded on two occasions with a fasting lipid profile.
- Age 8–10 years when LDL-C concentration >4.0 mmol/l, recorded on two occasions with a fasting lipid profile, in the presence of multiple ASCVD risk factors or family history of premature ASCVD.
- Age <8 years with an LDL-C concentration >4.9 mmol/l, recorded on two occasions.
- The use of treatment in this group does require specialist input as further study regarding the use is still needed.21
An LDL-C goal of <3.5 mmol/l or approximately 50% reduction may be considered in children/adolescents with no additional risk factors for ASCVD (e.g. diabetes, parental history of ASCVD in the second or third decade of life). Yearly non-fasting LDL-C levels in blood can be used to monitor those who have met LDL-C goal after initiation and titration of therapy, with no further dose change.19 Canadian Lipid Guidelines17 indicate LDL-C and non-HDL-C are interchangeable but non-HDL-C is preferable for non-fasting samples and if triglycerides > 1.5 mmol/L.
Lifestyle modifications discussed above remain the cornerstone of CVD prevention in both children and adolescents with FH and referral to a specialist for treatment decisions is recommended.1 The CCS recommends that children with HoFH are referred to a lipid specialist centre for cholesterol-lowering therapies when >15kg in weight.
Additional guidance on management of dyslipidemia in children and adolescence can be found here.22
Resources
- Canadian Lipid Clinics (June 2026)
- Genome BC has curated resources and links on FH.
- National Human Genome Research Institute has more on FH and genomics for the public and clinicians
- Infographic for individuals and families by the Canadian Cardiovascular Society and FHCanada
References
[1] Brunham LR, Ruel I, Aljenedil S, et al. Canadian Cardiovascular Society position statement on familial hypercholesterolemia: Update 2018. Can J Cardiol. 2018. 34(12):1553-1563.
[2] Bouhairie VE, Goldberg AC. Familial Hypercholesterolemia. Cardiol Clin. 2015. 33(2): 169-179.
[3] Vogt A. The genetics of familial hypercholesterolemia and emerging therapies. Appl Clin Genet. 2015. 8: 27-36.
[4] Beheshti SO, Madsen CM, Varbo A, Nordestgaard BG. Worldwide Prevalence of Familial Hypercholesterolemia: Meta-Analyses of 11 Million Subjects. J Am Coll Cardiol. 2020 May 26;75(20):2553-2566.
[5] Nordestgaard BG, Chapman MJ, Humphries SE, et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J. 2013. 34(45): 3478-3490a.
[6] Hartgers ML, Ray KK, Hovingh GK. New approaches in detection and treatment of familial hypercholesterolemia. Curr Cardio Rep. 2015. 17: 109.
[7] Hegele RA, Ban MR, Cao H, et al. Targeted next-generation sequencing in monogenic dyslipidemias. Curr Opin Lipidol. 2015; 26(2):103-13.
[8] Cuchel M, Raal FJ, Hegele RA, et al. 2023 Update on European Atherosclerosis Society Consensus Statement on Homozygous Familial Hypercholesterolaemia: new treatments and clinical guidance. Eur Heart J. 2023 Jul 1;44(25):2277-2291.
[9] Brunham L, Ruel I, Khoury E, et al. Familial hypercholesterolemia in Canada: initial results from the FH Canada National Registry. Atherosclerosis. 2018. 277: 419-424.
[10] Ruel I, Brisson D, Aljenedil S, et al. Simplified Canadian definition for familial hypercholesterolemia. Can J Cardiol. 2018. 34: 1210-1214.
[11] Turgeon RD, Barry AR, Pearson GJ. Familial hypercholesterolemia: review of diagnosis, screening, and treatment. Can Fam Physician. 2016. 62(1): 32-37.
[12] Austin MA, Hutter CM, Zimmern RL, et al. Genetic causes of monogenic heterozygous familial hypercholesterolemia: a HuGE prevalence review. Am J Epidemiol. 2004.160(5): 407-420.
[13] Knowles JW, Rader DJ, Khoury MJ. Cascade screening for familial hypercholesterolemia and the use of genetic testing. JAMA. 2017 Jul. 318(4): 381-382.
[14] Ned RM, Sijbrands EJ. Cascade screening for familial hypercholesterolemia (FH). PLoS Curr. 2011. 3: RRN1238.
[15] Sturm AC, Knowles JW, Gidding SS, et al. Clinical genetic testing for familial hypercholesterolemia: JACC scientific expert panel. J Am Coll Cardiol. 2018 Aug 7;72(6):662-680.
[16] Khoury M. Cascade screening in familial hypercholesterolemia: achieving buy-in and turning patients into partners. CJC Pediatr Congenit Heart Dis. 2023 Jun 20;2(5):219-221.
[17] Pearson GJ, Thanassoulis G, Anderson TJ, et al. 2021 Canadian Cardiovascular Society guidelines for the management of dyslipidemia for the prevention of cardiovascular disease in adults. Can J Cardiol. 2021 Aug;37(8):1129-1150.
[18] Kolber MR, Klarenbach S, Cauchon M, et al. PEER simplified lipid guideline 2023 update: prevention and management of cardiovascular disease in primary care. Can Fam Physician. 2023 Oct;69(10):675-686.
[19] Watts GF, Gidding SS, Hegele RA, et al. International Atherosclerosis Society guidance for implementing best practice in the care of familial hypercholesterolaemia. Nat Rev Cardiol. 2023 Dec;20(12):845-869.
[20] Paquette M, Bernard S, Cariou B, et al. A. Familial hypercholesterolemia-risk-score: a new score predicting cardiovascular events and cardiovascular mortality in familial hypercholesterolemia. Arterioscler Thromb Vasc Biol. 2021 Oct;41(10):2632-2640.
[21] Khoury M, Bigras JL, Cummings EA, et al. Dyslipidemia in children: diagnosis, evaluation, and management. Paediatr Child Health. 2026 Apr 30;31(4):392-399.
[22] Guirguis-Blake JM, Evans CV, Coppola EL, et al. Screening for lipid disorders in children and adolescents: updated evidence report and systematic review for the US Preventive Services Task Force. JAMA. 2023 Jul 18;330(3):261-274.
[23] Kramer AI, Christian S, Bartels K, et al. Genetic Testing for Familial Hypercholesterolemia: The Current State of Its Implementation in Canada. CJC Open. 2024 Aug 14;6(11):1395-1402
[24] Marchand M, Chen V, Trinder M, et al. Patient Perspectives Regarding Genetic Testing for Familial Hypercholesterolemia. CJC Open. 2021 Apr 8;3(5):557-564.
Additional Reading:
- Stein JH, Tattersall MC. Familial Hypercholesterolemia. JAMA. 2026 Jun 29. Epub ahead of print. PMID: 42371647.
- Ballantyne CM, Gellis L, Tardif JC, Banka P, Navar AM, Asprusten EA, Scott R, Stroes ESG, Froman S, Mendizabal G, Wang F, Catapano AL. Efficacy and Safety of Oral PCSK9 Inhibitor Enlicitide in Adults With Heterozygous Familial Hypercholesterolemia: A Randomized Clinical Trial. JAMA. 2026 Jan 13;335(2):129-139. PMID: 41206969
- Bellows BK, Zhang Y, Ruiz-Negrón N, Kazi DS, Khera AV, Woo JG, Urbina EM, Jacobs DR Jr, Allen NB, Wong JB, de Ferranti SD, Moran AE. Familial Hypercholesterolemia Screening in Childhood and Early Adulthood: A Cost-Effectiveness Study. JAMA. 2026 Jan 13;335(2):140-153. PMID: 41206967
- de Wert G, van El CG, Clarke A, Cordier C, Fellmann F, Genuardi M, Hentze S, Kayserili H, Macek M, MacLeod R, Melegh B, Mendes Á, Rial-Sebbag E, Stefánsdóttir V, Tranebjærg L, Ulph F, Forzano F. Cascade counselling and testing. Recommendations of the European Society of Human Genetics. Eur J Hum Genet. 2026 Feb;34(2):171-184. Epub 2025 Dec 14. PMID: 41392321
- Gao E, Brunham LR, Goldman RD. Advanced therapy in familial hypercholesterolemia. Can Fam Physician. 2026 Feb;72(2):106-108. PMID: 41679946
- Gillman MW. Childhood Cholesterol Screening Is Not Cost-Effective. JAMA. 2026 Jan 13;335(2):126-128. PMID: 41206971.
- O'Donoghue ML, Marston NA. Beyond Statins-Expanding the Arsenal of Lipid-Lowering Therapies. JAMA. 2026 Jan 13;335(2):124-125. PMID: 41206974
Authors: S Morrison MS CGC, JE Allanson MD FRCPC, RA Hegele MD FRCPC, S Walji MD CCFP and JC Carroll MD CCFP
GECKO Deep Dive is for educational purposes only and should not be used as a substitute for clinical judgement. GECKO aims to aid the practicing clinician by providing informed opinions regarding genetic services that have been developed in a rigorous and evidence-based manner. Physicians must use their own clinical judgment in addition to published articles and the information presented herein. GECKO assumes no responsibility or liability resulting from the use of information contained herein.

