![]()
Download the PDF here. Check out our FH point of care tools here and our more comprehensive overview, GECKO deep dive. Updated July 2026.
| Bottom line: Familial hypercholesterolemia (FH) is a common (~1/250) autosomal dominant condition that results in a 6- to 22-fold increase in premature cardiovascular disease (CVD) and death. Early diagnosis and treatment can normalize life expectancy. Key features of FH are elevated LDL-C ≥5 mmol/L, early onset CVD (<55 years in men, <65 years in women), cholesterol deposition in the tendons (xanthomata) and/or around the eyes (xanthelasma), arcus cornealis with onset <45 years, and family history of early onset CVD or hyperlipidemia requiring treatment. In Canada, a diagnosis of FH is typically based on an individual’s clinical presentation and history as outlined in the Canadian Cardiovascular Society algorithm. Genetic testing is not widely clinically available in Canada with some exceptions. A clinical diagnosis guides treatment and screening of family members. Once a person is diagnosed with FH, cascade screening of family members using measurement of LDL-C levels and/or genetic testing is recommended. This enables early identification and treatment of at-risk individuals, with statins as first-line treatment. |
What is Familial Hypercholesterolemia?
Familial hypercholesterolemia (FH) is an autosomal dominant genetic condition where the uptake of low-density lipoprotein cholesterol (LDL-C) into cells is either decreased or inhibited. This results in lifetime exposure to very high levels of LDL-C. FH is the most common genetic disorder causing premature cardiovascular disease (CVD) and death in both men and women. FH is both underdiagnosed and undertreated worldwide despite the knowledge that early diagnosis and treatment can normalize life expectancy.1-3 It is estimated that roughly 1 in 250 Canadians has FH, and that only about 10% have been identified.1 The global prevalence of FH in the general population is about 1 in 300 and is 10- to 20-fold more common among individuals with ischemic heart disease, particularly premature ischemic heart disease.4
Most cases (up to 80%) of familial hypercholesterolemia (FH) are caused by pathogenic/likely pathogenic (P/LP) variants (what used to be called mutations) in the LDL receptor gene LDLR, in which > 3000 different P/LP variants have been identified.2,5,6
How is familial hypercholesterolemia diagnosed?
The Canadian Cardiovascular Society (CCS) recommends the use of the Canadian diagnostic criteria for FH proposed by the Familial Hypercholesterolemia Canada (FHCanada) network (Figure 1).10 Genetic testing is a key approach to the diagnosis of definite FH. While clinical criteria can be used for diagnosis, there are limitations to using the classical presentation since few affected persons will exhibit physical findings (e.g. xanthomas, xanthelasmas) at the time of testing.15 Additionally, there are limitations to use of family history of cardiovascular disease in FH diagnosis. Screening for FH based on family history alone has been shown to miss 30-60% of cases.16 Genetic testing is not necessary for diagnosis and is not yet routinely clinically available in most of Canada.
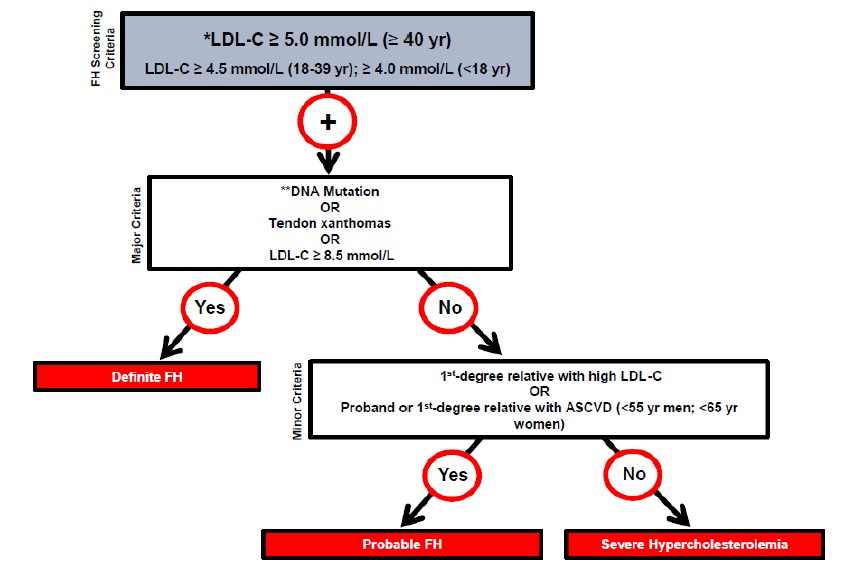
Figure 1. Canadian criteria for the clinical diagnosis of familial hypercholesterolemia (FH). From Ruel I et al, 201810. Reprinted with permission under the CC BY-NC-ND license https://creativecommons.org/licenses/by-nc-nd/4.0/. DOI: 10.1016/j.cjca.2018.05.015
ASCVD: atherosclerotic cardiovascular disease; LDL-C: low-density lipoprotein cholesterol. * Secondary causes of high LDL-C should be ruled out (severe or untreated hypothyroidism, nephrotic syndrome, hepatic disease [biliary cirrhosis], medication, especially antiretroviral agents) ** DNA mutation refers to the presence of a known FH-causing variant in a FH gene in the individual or a first-degree relative. FH diagnosis in a patient with a P/LP variant but normal LDL-C levels is unclear. Yearly follow-up of the individual is suggested, and cascade screening of family members should be initiated.
The LDL-C levels cited are untreated levels. For a patient who is on lipid lowering medications, the CardioRisk CalculatorTM app has a validated algorithm to assign a baseline value.
How to order genetic testing for Familial Hypercholesterolemia?
While genetic testing is not necessary for diagnosis It can confirm a diagnosis of FH, identify at-risk relatives early through cascade testing, support cardiovascular risk assessment, and has shown to improve treatment uptake, adherence, and LDL cholesterol reduction.22
Genetic testing for FH is not available throughout Canada and some provinces have ordering restrictions.22 Contact your local genetics centre for support. See below for how genetic testing can be accessed in your province.
- Testing must be ordered by geneticist in Manitoba and Newfoundland and Labrador.
- In addition to geneticists, specialist physicians in endocrinology, cardiology and internal medicine can order genetic testing for FH in Alberta, Saskatchewan and Nova Scotia.
- Genetic testing for FH is not yet clinically available in British Colombia.
- In Ontario and Quebec, genetic testing can be ordered by any physician and does not require a referral for a specialist assessment
Genetic testing in Québec, select the laboratory closest to you: (July 2026)
- The Core Molecular Diagnostic Laboratory at the McGill University Health Centre: Core Molecular Diagnostics Requisition (DM 5891)
- CHU Sainte Justine Molecular Laboratory
Ontario, select the laboratory closest to you: (July 2026, alternately you can use the Ontario Genetic Test Directory to search):
- London Health Sciences Centre (LHSC Requisition) molecular laboratory
- Trillium Health Partners – Credit Valley Site (THP Requisition)
- Hamilton Regional Laboratory Medicine Program (Requisition)
Cascade Screening For Family Members
The most cost-effective approach for identification of new FH cases is cascade screening of family members of the first individual with a confirmed diagnosis, known as the index case.4,11,13 Data from the UK have shown that cascade screening reduces the average age at which an individual is diagnosed and results in an increased number of individuals who are treated with statins and have subsequent lowered lipid levels.14
The Canadian Cardiovascular Society (CCS) recommends screening of first-degree relatives of the index case.1 Screening can include lipid profiles of relatives and/or genetic testing for a known familial P/LP variant, when available. Each newly diagnosed individual becomes a new index case and cascade screening of relatives continues.
When using a genetic testing approach, testing relatives for a known familial P/LP variant, positive results will identify at-risk relatives and negative results would reassure those at population risk. When ordering genetic testing for relatives it is important to include documentation of the familial genetic variant either with a molecular report or a family letter. This ensures accurate interpretation of testing.
Surveillance and Management
adults
The use of conventional cardiovascular risk calculators in individuals with FH is not recommended as these greatly underestimate lifetime CVD risk.1,2, 19 FH-specific cardiovascular risk calculators (e.g. the FH Risk Score20, SAFEHEART 19) should be considered to assess the risk of ASCVD in those with FH. Routine assessment and stratification of the risk of ASCVD in all patients with FH should be used to guide personalized treatment and management.19 Referral to specialist for risk stratification can be considered.
Those with homozygous status (HoFH, two pathogenic/likely pathogenic (P/LP) variants in an FH gene) should be referred to a specialized lipid centre.1
A genetic diagnosis of FH in a person with a P/LP variant but normal LDL-C levels is unclear. Yearly follow-up of the individual is suggested, and cascade screening of family members should be initiated.1
For management and screening recommendations for those individuals with FH, see the 2018 CCS Position Statement and the more recently published evidence-based guidelines by The International Atherosclerosis Society.22
Pharmaceuticals
Statins are the drug class of choice for individuals with one P/LP variant in a FH gene (HeFH). Observational studies have shown a dramatic decrease in cardiac events in statin-treated individuals with FH.1 LDL-C should be lowered as fast and as far as possible.3 The CCS recommends a >50% reduction of LDL-C as primary prevention and that an ideal goal of LDL-C <1.8 mmol/L is recommended for secondary prevention.17 Non-fasting lipid profiles should be used to monitor treatment in those whose treatment is stable.20
For the most recent recommendations on management and treatment of individuals with HoFH please see Cuchel et al. 2023.8
Lifestyle
All families with FH (including children and adolescents) should be counselled about the importance of lifestyle modification and heart healthy behaviour1-3,17,19 such as smoking cessation and avoidance of passive smoking, diet (e.g. high in fibre, low in trans and saturated fatty acids), exercise, stress reduction, maintenance of ideal body weight.
Pregnancy
For most persons assigned female at birth who are of reproductive age, an effective birth control method is recommended with discontinuation of statin therapy ideally 3 months prior to planned pregnancy or at the time of a positive pregnancy test.1,20,21 A pregnant person with FH and additional risk factors, e.g. established ASCVD, should be referred to a specialty lipid clinic for further treatment advice.
Children /Adolescents
The CCS and Canadian Pediatric Cardiology Association recommend universal lipid screening (fasting or non-fasting, non-HDL-C or LDL-C) be performed after 2 years of age and within the first decade of life.21 Many clinicians, however, are not routinely implementing this practice due to controversies around the utility of such a test. Selective screening at any time can be considered when there is a positive family history of premature CVD or dyslipidemia, or other cardiovascular risk factors.21 Reverse cascade screening of parents is recommended when a child is found to have FH.1,21
The ideal age to begin treatment is between 8 and 12 years of age based on current randomized control trials.21 Pharmacological treatment can be considered, incorporating clinical judgement, family and patient preferences.1,19,21
Lifestyle modifications discussed above remain the cornerstone of CVD prevention in both children and adolescents with FH and referral to a specialist for treatment decisions is recommended.1 The CCS recommends that children with HoFH are referred to a lipid specialist centre for cholesterol-lowering therapies when >15kg in weight.
Additional guidance on management of dyslipidemia in children and adolescence can be found here.21
Resources, References and Additional Reading
See the GECKO deep dive
Authors: S Morrison MS CGC, JE Allanson MD FRCPC, RA Hegele MD FRCPC, S Walji MD CCFP and JC Carroll MD CCFP
GECKO on the run is for educational purposes only and should not be used as a substitute for clinical judgement. GECKO aims to aid the practicing clinician by providing informed opinions regarding genetic services that have been developed in a rigorous and evidence-based manner. Physicians must use their own clinical judgment in addition to published articles and the information presented herein. GECKO assumes no responsibility or liability resulting from the use of information contained herein.

