
Download the PDF here also available is a point of care tool to triage those who would benefit from genetic referral.
Click here for a case-based education module.
Lynch syndrome (LS), also known as Hereditary Non-Polyposis Colorectal Cancer (HNPCC), is the most common hereditary colorectal (CRC) cancer predisposition syndrome. It is an autosomal dominant condition that results in an increased lifetime risk of colorectal cancer (CRC) in addition to other cancers. Individuals at high or intermediate risk of LS should be referred for a genetic consultation for consideration of genetic testing. The results of genetic testing can lead to appropriate surveillance and improved outcomes for both affected individuals and their family members. Conversations between patients and their healthcare providers are strong drivers of screening participation.
Updated January 2019
WHAT IS LYNCH SYNDROME?
Lynch syndrome (LS), also known as Hereditary Non-Polyposis Colorectal Cancer (HNPCC), is the most common inherited cause of CRC.3 LS accounts for about 0.7-3.6% of cases of CRC.4 LS is caused by an inherited mutation in one of four mismatch repair (MMR) genes (MLH1, MSH2, MSH6, PMS2) or in EPCAM, a gene that plays a role in MSH6 gene inactivation. Having an LS mutation results in an increased lifetime risk of CRC and other LS –related cancers (see Box 1).

PERSONAL HISTORY RED FLAGS TO CONSIDER GENETIC TESTING OR GENETIC CONSULTATION
These are general guidelines to identify patients at high risk for LS. You should check with your local genetics centre or hereditary cancer program for more specific details. Consider referring your patient if he/she has:
- An early age of CRC diagnosis (<50 years). Diagnosis under age 35 is more likely to be LS
- An early age of endometrial cancer diagnosis (<50 years)
- Multiple primary LS-related cancer diagnoses, regardless of age
- A CRC diagnosis and one or more 1st degree relatives with a LS-related cancer, with one of the cancers being diagnosed <50 years
- A CRC diagnosis and two or more 1st or 2nd degree relatives with LS- related cancers regardless of age
- A CRC diagnosis <60 years with histological features suspicious for LS (excess infiltrating lymphocytes, mucinous/signet cell features, Crohn’s-like reaction), particularly when primary tumour is right-sided
FAMILY HISTORY RED FLAGS TO CONSIDER GENETIC CONSULTATION
You should consider referring your patient to your local genetics centre or hereditary cancer program for further assessment if he/she is at high risk for hereditary CRC syndrome.
A patient is considered to be at high risk for LS syndrome if he/she
- Has a known LS causing mutation in the family
Or if he/she meets the revised Amsterdam criteria, meaning he/she:
Has at least three relatives with a cancer associated with LS (Box 1); the following criteria should also be present:
- One must be a first degree relative of the other two;
- At least two successive generations must be affected (autosomal dominant inheritance);
- At least one relative with LS-related cancer should be diagnosed before age 50;
Tumour pathology should be verified when possible and other CRC syndromes should be ruled out
If your patient does not have cancer, genetic testing of a relative with cancer may be recommended as a first step.
HOW IS GENETIC TESTING DONE?
Ideally testing begins with immunohistochemical (IHC) analysis of a CRC tumour for the protein products of the LS genes (MLH1, MSH2, MSH6, and PMS2). If IHC analysis reveals a protein to be deficient, genetic testing can be offered to the affected individual and performed on a blood sample. If IHC analysis does not clearly show protein deficiency, the next step is often microsatellite instability (MSI) testing of the tumour sample. If MSI is stable or low, no further testing is indicated. If MSI is high, genetic testing can be offered to the affected individual. Some centres will arrange IHC or MSI alone; others will carry out both tests at the same time.
WHAT DOES THE GENETIC TEST RESULT MEAN?
If your patient has been found to carry a mutation in a LS gene, a positive result, they have an increased lifetime risk to develop certain cancers (Table 1 and Box 1). Variable expressivity and incomplete penetrance must also be kept in mind. This result also means that family members are at risk of carrying the same mutation and of having similar cancer risks.
If a mutation is not identified in someone from a family with a known mutation, this is a true negative result. You can provide reassurance to your patient. These individuals may still have modified screening recommendations based on their family history. Consult your local genetics centre or hereditary cancer program.
If a mutation is not identified in an affected patient who has no known familial mutation this result is uninformative. In this case, the diagnosis of LS is not confirmed or ruled out, especially in families with a strong history of CRC.
Table 1. Significant lifetime cancer risks for individuals who have inherited a mutation in the LS genes, MLH1 and MSH2, as compared to the general population. Risks for other LS genes are lower, and can be found in the GEC-KO Messenger (in addition to other LS-related cancer risks).
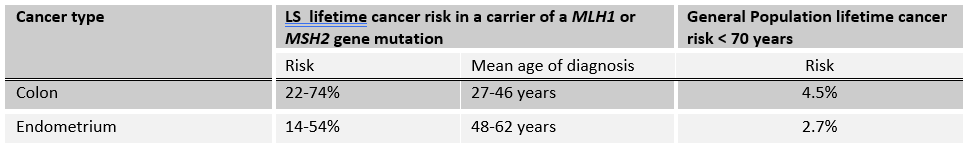
CLINICAL UTILITY
Identification of a gene mutation can lead to appropriate surveillance and improved outcomes for affected individuals. It can also guide testing of at-risk family members.
SCREENING AND SURVEILLANCE
In general, for individuals at high risk for CRC (carriers of a mutation in a LS gene and their first degree relatives who have not yet had genetic testing) screening recommendations are as follows:
Colorectal Cancer: Colonoscopy every 1-2 years beginning between ages 20 and 25 or 2-5 years prior to the earliest diagnosis if that diagnosis was made before age 25 years, whichever is earlier.1
Endometrial and Ovarian cancer: Screening for endometrial or ovarian cancer may include transvaginal ultrasound and endometrial biopsy, however, there is little evidence of the effectiveness of these tests. 2
Individuals who have tested negative for a known familial LS gene should follow provincial guidelines for population risk CRC screening, i.e. Fecal Occult Blood Test every two years from age 50. For those individuals who have a family history of CRC unrelated to the mutation in their family (i.e. on the other side of the family), screening recommendations would be based on the family history. Consult your local genetics centre or hereditary cancer program.
CRC screening for individuals at increased risk is dependent on family history. For a person with a3:
- 1st degree relative with CRC diagnosis at any age → Colonoscopy at age 40 or 10 years younger than the youngest CRC diagnosis, repeat 5 yearly
- 2nd degree relative with CRC diagnosis <50 years → Colonoscopy at age 50, repeat 5-10 yearly
See www.geneticseducation.ca for the full GEC-KO Messenger on LS for more details
References
[1] Referenced with permission from the NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for Genetic/Familial High-Risk Assessment: Colorectal V.1.2018 © National Comprehensive Cancer Network, Inc 2018. All rights reserved. [Accessed January 31, 2019]. To view the most recent and complete version of the guideline, go online to www.nccn.org
[2] Vasen HFA, Blanco I, Akhtan-Collan K, Gopie JP, Alonso A, Aretz S, et al. Revised guidelines for the clinical management of Lynch syndrome (HNPCC): recommendations by a group of European experts. Gut. 2013; 62:812–823.
[3] Referenced with permission from the NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for Colorectal Cancer Screening V.1.2018 © National Comprehensive Cancer Network, Inc 2018. All rights reserved. [Accessed January 31, 2019]. To view the most recent and complete version of the guideline, go online to www.nccn.org
Authors: S Morrison MS CGC, JE Allanson MD FRCPC, E Tomiak MD FRCPC, K Semotiuk MS (C)CGC and JC Carroll MD CCFP
Updated by the GECKO team: JC Carroll MD CCFP, JE Allanson MD FRCPC FCCMG, S Yusuf MS CGC
Disclaimer:
· GECKO is an independent not-for-profit program that does not accept support from commercial or non-academic entities.
· GECKO aims to aid the practicing non-genetics clinician by providing informed resources regarding genetic/genomic conditions, services and technologies that have been developed in a rigorous and evidence-based manner with periodic updating. The content on the GECKO site is for educational purposes only. No resource should be used as a substitute for clinical judgement. GECKO assumes no responsibility or liability resulting from the use of information contained herein.
· All clinicians using this site are encouraged to consult local genetics clinics, medical geneticists, or specialists for clarification of questions that arise relating to specific patient problems.
· All patients should seek the advice of their own physician or other qualified clinician regarding any medical questions or conditions.
· External links are selected and reviewed at the time a page is published. However, GECKO is not responsible for the content of external websites. The inclusion of a link to an external website from GECKO should not be understood to be an endorsement of that website or the site’s owners (or their products/services).
· We strive to provide accurate, timely, unbiased, and up-to-date information on this site, and make every attempt to ensure the integrity of the site. However, it is possible that the information contained here may contain inaccuracies or errors for which neither GECKO nor its funding agencies assume responsibility.

